SAFETY OF MEDICAL DEVICES
Improving the mechanisms for collecting and analyzing data on the safety of medical devices
Author information
1 — Federal State Budgetary Educational Institution of Higher Education “Saint-Petersburg State University”, 7/9, Universitetskaya nab., St. Petersburg, 199034, Russian Federation.
ORCID: https://orcid.org/0000-0002-7765-4876
Published: 08.12.2023
Regularly appearing data on serious incidents related to the use of medical devices make it necessary to improve the mechanisms for monitoring their safety in order to prevent harm to the life and health of citizens and medical workers. The article presents the main trends in regulatory regulation of monitoring the safety of medical devices: the author analyzes the main international and national regulations, studied information letters about medical devices posted on the official website of Roszdravnadzor for the period 2015–2022. The growth and globalization of the medical devices market, the need to ensure the safety of their use determine the general vector of development of regulation at all levels, including the legislation of the Russian Federation - increased attention to the collection and analysis of safety data at the post-registration stage with the expansion of functions and increased responsibility of manufacturers, the formation of effective monitoring mechanisms safety of medical devices.
Keywords: Medical devices, monitoring the safety of medical devices, regulatory acts, safety of medical practice
Background.
В последние десятилетия на фоне бурного развития технологий регулярно публикуются данные о наступлении неблагоприятных событий при применении медицинских изделий (МИ) [1][2]. Очевидно, что в условиях разработки и внедрения в медицинскую практику новых МИ, постоянной модификации имеющихся возрастает актуальность накопления опыта их применения, в том числе негативного, предотвращения причинения вреда жизни и здоровью пользователей (граждан и медицинских работников). Ключевым инструментом в минимизации неблагоприятных событий является мониторинг безопасности МИ. Его цель – выявление и предотвращение побочных действий, не указанных в эксплуатационной документации, нежелательных реакций при применении МИ, особенностей взаимодействия МИ между собой, фактов и обстоятельств, создающих угрозу жизни и здоровью пользователей при применении МИ.
Ускорение и упрощение процедуры регистрации для отдельных групп МИ, снижение административной нагрузки на субъекты обращения МИ (внедрение Росздравнадзором риск-ориентированного подхода к проведению контрольных мероприятий, смещение акцента с проверок на профилактику правонарушений) в последние годы накладывает на их руководителей дополнительную ответственность за организацию добросовестного выполнения сотрудниками обязанностей в рамках мониторинга безопасности МИ, повышает значимость работы в данном направлении.
Цель исследования
Выявить основные тенденции нормативного регулирования проведения мониторинга безопасности МИ, оценить их влияние на количество выявляемых Росздравнадзором новых данных по безопасности.
Материалы и методы
Проанализированы основные международные и национальные нормативные акты, регулирующие проведение мониторинга безопасности МИ. Изучены информационные письма о МИ, размещенные на официальном сайте Росздравнадзора1, опубликованные в 2015–2022 гг. Из общего числа записей о МИ за 7 лет (7703 ед.) выделены сообщения о новых данных по безопасности.
В исследовании применялись методы системного и логического анализа, сравнения и описания. Статистический анализ полученных данных осуществлялся с помощью классических процедур и методов: данные представлены в виде абсолютных значений и долей от целого. Сравнение долей между несвязанными выборками осуществлялось критерием хи-квадрат.
Results.
Глобальное регулирование сбора и анализа данных по безопасности МИ. В условиях роста и глобализации рынка МИ ключевой задачей становится оптимизация взаимодействия всех субъектов их обращения. Формирование общих подходов к регулированию отношений в данной сфере, выработку единых определений, стандартов на всех этапах жизненного цикла МИ, в том числе на этапе применения, в последние десятилетия активно осуществляют Всемирная организация здравоохранения (ВОЗ), Международный форум по регулированию медицинских изделий (IMDRF; International Medical Device Regulators Forum).
Так, в 2017 году опубликована глобальная рамочная модель ВОЗ по регулированию МИ2. Модель содержит правила надлежащей регуляторной практики, сформированные на основе опыта стран с развитым регулированием (США, ЕС, Швейцарии и других). В документе описаны стадии жизненного цикла МИ, сформирован понятийный аппарат, единые подходы к основным проблемам обращения, в том числе на пострегистрационном (пострыночном) этапе жизненного цикла МИ. Основным трендом становится уменьшение общего времени, затрачиваемого на внедрение в медицинскую практику безопасных и востребованных МИ, с одновременным укреплением контроля на пострегистрационном этапе, повышением эффективности сбора и анализа информации от пользователей, а в случае выявления дефектов (неисправностей) – принятия необходимых мер для недопущения их повторения. В рамках данного подхода в 2021 году ВОЗ издано «Руководство по пострегистрационному надзору за медицинскими изделиями, в том числе используемыми для диагностики in vitro»3. В руководстве ВОЗ под пострегистрационным надзором понимается комплекс мероприятий, проводимых производителями для сбора информации о применении МИ, анализ опыта их использования, а также для определения необходимости принятия тех или иных мер, направленных на повышение безопасности. Отмечается важность проведения производителями МИ анализа информации, получаемой в рамках «обратной связи», в первую очередь от медицинских организаций, а также использование механизмов упреждающего (проактивного) контроля.
В соответствии со стратегией IMDRF4, членом Руководящего комитета которого является Российская Федерация, опубликованной в сентябре 2020 года, к приоритетным направлениям развития регулирования пострегистрационного этапа жизненного цикла МИ относятся меры по совершенствованию мониторинга безопасности МИ:
- проработка терминов и кодов, используемых для сообщения о неблагоприятных событиях при применении МИ;
- разработка рекомендаций для создания и использования уникальных идентификаторов МИ (UDI) для эффективного сбора информации в рамках мониторинга безопасности МИ.
Одно из направлений глобального регулирования – создание единой номенклатуры (классификатора) МИ, которая бы использовалась большинством стран, – является крайне важным, в том числе для целей сбора и анализа данных по безопасности МИ, так как позволит систематизировать и более эффективно использовать разрозненные в настоящее время сведения [3].
Регулирование в рамках Евразийского экономического союза. Ведущими экономическими блоками, такими как Евразийский экономический союз (ЕАЭС), Европейский Союз (ЕС) и другими активно принимаются правила в сфере обращения МИ, осуществляется гармонизация требований законодательства государств-членов.
В целях создания единого рынка МИ в рамках ЕАЭС с 2012 года при Коллегии Евразийской экономической комиссии сформирована рабочая группа для подготовки общих подходов к регулированию обращения МИ. Основополагающим документом стало Соглашение о единых принципах и правилах обращения медицинских изделий (изделий медицинского назначения и медицинской техники) в рамках Евразийского экономического союза, заключенное 23 декабря 2014 года.5
В рамках указанного соглашения Советом Евразийской экономической комиссии, Коллегией Евразийской экономической комиссии в настоящее время принято несколько десятков решений об утверждении требований, критериев, правил в сфере обращения МИ, в том числе регулирующих проведение мониторинга безопасности МИ.
Проведение мониторинга безопасности МИ, зарегистрированных в соответствии с актами, составляющими право ЕАЭС, и находящихся в обращении на территории РФ, осуществляется в соответствии с законодательством ЕАЭС, в частности с Решением Коллегии Евразийской экономической комиссии от 22.12.2015 № 174 «Об утверждении Правил проведения мониторинга безопасности, качества и эффективности медицинских изделий»6. Документом определено, что целями мониторинга являются обеспечение безопасности пользователей, сохранение и укрепление здоровья населения, повышение качества оказания медицинской помощи, выявление и предотвращение побочных действий и нежелательных реакций, не указанных в инструкции по применению или руководстве по эксплуатации МИ, неблагоприятных событий (инцидентов), обращения МИ, не соответствующих утверждаемым Евразийской экономической комиссией общим требованиям безопасности и эффективности МИ, требованиям к их маркировке и эксплуатационной документации на них. Даны определения целому ряду понятий, таким как «неблагоприятное событие (инцидент)», «нежелательное событие», «серьезная угроза здоровью» и другим. Установлены принципы взаимодействия субъектов обращения МИ с уполномоченными органами государств-членов ЕАЭС, уполномоченных органов государств-членов ЕАЭС друг с другом в рамках мониторинга.
Уполномоченными органами государств-членов ЕАЭС осуществляется информационное взаимодействие в единой информационной базе данных мониторинга безопасности, качества и эффективности МИ, формирование и ведение которой в электронном виде осуществляется Евразийской экономической комиссией7. Такое взаимодействие направлено на:
- снижение издержек, связанных с обменом информацией о результатах мониторинга безопасности, качества и эффективности МИ, за счет создания общего информационного пространства в сфере обращения МИ;
- предотвращение обращения на территории ЕАЭС МИ, не соответствующих требованиям безопасности, качества и эффективности, за счет оперативного представления участникам общего процесса актуальной информации посредством интегрированной информационной системы внешней и взаимной торговли.
Российская Федерация, играя ведущую роль в разработке механизмов мониторинга безопасности, качества и эффективности МИ в ЕАЭС, активно совершенствует и гармонизирует с требованиями ЕАЭС национальное законодательство.
Регулирование в Российской Федерации. Основные принципы проведения мониторинга безопасности МИ в РФ определены в статье 96 Федерального закона от 21.11.2011 № 323-ФЗ «Об основах охраны здоровья граждан в Российской Федерации»8.
Мониторинг включает в себя сбор и анализ информации о неблагоприятных событиях, связанных с МИ, находящихся в обращении на территории РФ, на всех этапах обращения МИ, выявленных как на территории РФ, так и других государств, принятие соответствующих решений уполномоченным органом (Росздравнадзором), которые размещаются на его сайте.
Все субъекты обращения МИ обязаны сообщать в Росздравнадзор о побочных действиях, не указанных в инструкции по применению или руководстве по эксплуатации МИ, нежелательных реакциях при его применении, особенностях взаимодействия МИ между собой, фактах и обстоятельствах, создающих угрозу жизни и здоровью граждан и медицинских работников при применении и эксплуатации МИ в течение двадцати рабочих дней со дня выявления. Невыполнение указанной обязанности влечет привлечение к административной ответственности.
В рамках мониторинга в каждом конкретном случае Росздравнадзору при участии производителя и экспертного учреждения необходимо установить наличие (отсутствие) причинно-следственной связи между применением МИ и развитием выявленных неблагоприятных событий, принять в случае необходимости меры по предотвращению причинения вреда жизни и здоровью граждан и медицинских работников (например, приостановление применения МИ (модели (варианта) исполнения, партии или серии МИ). Далее, в зависимости от результатов контрольных и других мероприятий, производителю может быть предложено разработать и принять комплекс мер, направленных на исключение возможности в дальнейшем возникновения подобных событий.
В 2020–2021 гг. усовершенствованы механизмы мониторинга безопасности МИ, в частности, определены этапы, сроки, регламентированы действия участников, в первую очередь, производителей.
Можно выделить следующие основные нововведения:
- утверждена классификация неблагоприятных событий, связанных с обращением МИ9;
- введена обязанность для производителя (его уполномоченного представителя) для МИ класса потенциального риска применения 3, а также МИ, имплантируемых в организм человека класса потенциального риска применения 2б, проводить мониторинг безопасности и клинической эффективности после регистрации (клинический мониторинг) с предоставлением в Росздравнадзор отчетов10;
- расширен список источников получения Росздравнадзором информации для осуществления мониторинга безопасности МИ (информационные системы в сфере здравоохранения, интернет-сайты зарубежных регуляторных органов в сфере обращения МИ, представляемые производителями результаты клинического мониторинга МИ класса потенциального риска применения 3, а также имплантируемых в организм человека МИ классов 2)11;
- определены обязанности производителей МИ по своевременному предоставлению информации (уведомлений по безопасности МИ10, программ корректирующих мероприятий11) в доступной форме для неограниченного круга пользователей;
- к обязанностям медицинских организаций в рамках мониторинга отнесено направление сообщений о неблагоприятных событиях не только в Росздравнадзор, но и производителю (его уполномоченному представителю);
- создание информационной системы «Регистр пациентов с имплантированными медицинскими изделиями».
Анализ сообщений о новых данных по безопасности МИ за 2015–2022 гг. показывает, что их количество за указанный период возросло, существенно (р < 0,05) увеличилась их доля в общей структуре информационных писем о МИ Росздравнадзора (рисунок), что свидетельствует об эффективности механизмов мониторинга безопасности МИ.
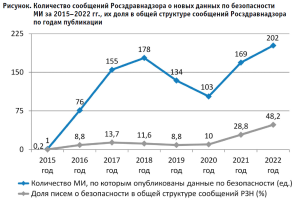
Conclusion.
Рост и глобализация рынка МИ, необходимость обеспечения безопасности их применения обусловливают общий вектор развития регулирования на всех уровнях, включая законодательство Российской Федерации, – усиление внимания к сбору и анализу данных по безопасности на пострегистрационном этапе с расширением функций и повышением ответственности производителей, формирование эффективных механизмов мониторинга безопасности МИ.
Отмечается рост числа сообщений Росздравнадзора о выявляемых данных по безопасности МИ, их доли в общей структуре информационных писем Росздравнадзора за последние 7 лет, что способствует повышению их безопасного применения.
Дальнейшее совершенствование нормативной базы, направленное на предупреждение, выявление и предотвращение рисков причинения вреда жизни и здоровью граждан при применении МИ, сообщение медицинскими организациями о неблагоприятных событиях при оказании медицинской помощи, использование ими новых данных по безопасности применительно к имеющимся МИ остаются важнейшими задачами в рамках повышения безопасности медицинской деятельности.
_____________________________________________________________________
1 URL: https://roszdravnadzor.gov.ru.
2 Глобальная рамочная модель ВОЗ по регулированию медицинских изделий, в том числе медицинских изделий для диагностики in vitro: офиц. текст [Электронный ресурс]. – URL: https://apps.who.int/iris/handle/10665/352256?locale-attribute=ru& (дата обращения: 01.03.2023).
3 Руководство по пострегистрационному надзору за медицинскими изделиями, в том числе используемыми для диагностики in vitro [Guidance for post-market surveillance and market surveillance of medical devices, including in vitro diagnostics]. Женева: Всемирная организация здравоохранения; 2021 г. Лицензия: CC BY-NC-SA 3.0 IGO.
4 IMDRF Strategic Plan 2021-2025. 25 September 2020 [Электронный ресурс]. – URL: http://www.imdrf.org/docs/imdrf/final/procedural/imdrf-proc-n39-strategic-plan-200925.pdf (дата обращения: 01.03.2021).
5 Соглашение о единых принципах и правилах обращения медицинских изделий (изделий медицинского назначения и медицинской техники) в рамках Евразийского экономического союза. (Заключено в г. Москве 23.12.2014).
6 Решение Коллегии Евразийской экономической комиссии от 22.12.2015 № 174 «Об утверждении Правил проведения мониторинга безопасности, качества и эффективности медицинских изделий»: офиц. текст [Электронный ресурс]. – URL: https://base.garant.ru/71296496/(дата обращения: 11.03.2023).
7 Распоряжение Коллегии Евразийской экономической комиссии от 12.11.2018 № 177 «О введении в действие общего процесса “Формирование, ведение и использование единой информационной базы данных мониторинга безопасности, качества и эффективности медицинских изделий”»: офиц. текст [Электронный ресурс]. – URL: http://www.consultant.ru/document/cons_doc_LAW_311222/ (дата обращения: 06.03.2023).
8 Федеральный закон от 21.11.2011 № 323-ФЗ «Об основах охраны здоровья граждан в Российской Федерации»: офиц. Текст [Электронный ресурс]. – URL: https://base.garant.ru/12191967/aa57c2128bfc4a5ccfe4faafe888d6af/ (дата обращения: 06.03.2023).
9 Приказ Росздравнадзора от 20.05.2021 № 4513 «Об утверждении классификации неблагоприятных событий, связанных с обращением медицинских изделий»: офиц. текст [Электронный ресурс]. – URL: http://ivo.garant.ru/#%2Fdocument%2F400884307%2Fparagraph%2F59%3A4 (дата обращения: 11.03.2023).
10 Приказ Минздрава России от 19.10.2020 № 1113н «Об утверждении Порядка сообщения субъектами обращения медицинских изделий обо всех случаях выявления побочных действий, не указанных в инструкции по применению или руководстве по эксплуатации медицинского изделия, о нежелательных реакциях при его применении, об особенностях взаимодействия медицинских изделий между собой, о фактах и об обстоятельствах, создающих угрозу жизни и здоровью граждан и медицинских работников при применении и эксплуатации медицинских изделий»: офиц. текст [Электронный ресурс]. – URL: https://base.garant.ru/75011335/ (дата обращения: 06.03.2023).
11 Приказ Минздрава России от 15.09.2020 № 980н «Об утверждении Порядка осуществления мониторинга безопасности медицинских изделий»: офиц. текст [Электронный ресурс]. – URL: https://base.garant.ru/74848249/#block_1000 (дата обращения: 06.03.2023).
- Hauser R.G., Abdelhadi R., McGriff D., Retel L.K. Deaths caused by the failure of Riata and Riata ST implantable cardioverterdefibrillator leads // Heart Rhythm, 2012; 9: 1227–1235.
- Collett D.J., Rakhorst H., Lennox P., Magnusson M., Cooter R., Deva A.K. Current Risk Estimate of Breast Implant-Associated Anaplastic Large Cell Lymphoma in Textured Breast Implants // Plast Reconstr Surg. 2019 Mar; 143(3S A Review of Breast Implant-Associated Anaplastic Large Cell Lymphoma): 30S-40S.
- Issues of registration of medical devices and monitoring of their safety during medical use in various countries / E.M. Astapenko, A.A. Kamaletdinova, A.V. Kolokolov, D.V. Ognerubov // Bioeconomics: doctrine, legislation, practice: monograph. – Moscow : Prospect Limited Liability Company, 2021. – pp. 271–311. (in Russian).