REGISTRATION OF MEDICAL DEVICES
Features of registration of medical devices on the territory of the Russian Federation during the COVID-19 coronavirus infection pandemic
Author information
1 — Federal Service for Surveillance in Healthcare, 4, bld. 1, Slavyanskaya Square, Moscow, 109074, Russian Federation.
2 — Federal Service for Surveillance in Healthcare, 4, bld. 1, Slavyanskaya Square, Moscow, 109074, Russian Federation.
Published: 25.06.2021
The article discusses issues related to the features of state registration of medical devices during a pandemic of a new coronavirus infection. The key points that applicants should pay attention to when preparing documents for state registration of medical devices are reflected.
Keywords: medical devices, tests, features of registration, list of medical devices, simplified scheme, expedited registration
В целях организации мер по профилактике и недопущению распространения новой коронавирусной инфекции в 2020 г. было организовано ускоренное обеспечение граждан Российской Федерации, а также медицинских работников средствами защиты, реагентами in vitro, медицинской техникой и другими изделиями, предназначенными для выявления, лечения и профилактики COVID-19. Правительством Российской Федерации были изданы нормативные акты, позволяющие ускорить государственную регистрацию медицинских изделий. Постановлением Правительства Российской Федерации от 18.03.2020 № 299 «О внесении изменений в Правила государственной регистрации медицинских изделий» (далее – Постановление Правительства РФ № 299) введен особый порядок государственной регистрации медицинских изделий с низкой степенью потенциального риска применения, которые включены в перечень (далее – Перечень), представленный в приложении к Правилам государственной регистрации медицинских изделий, утвержденным Постановлением Правительства Российской Федерации от 27.12.2012 № 1416 (далее – Правила), заключающийся в предоставлении упрощенного пакета документов и существенного сокращения сроков регистрации.
Упрощенный порядок государственной регистрации медицинских изделий с низкой степенью потенциального риска применения
Государственная регистрация медицинского изделия, включенного в Перечень, проводится в срок, не превышающий 8 рабочих дней после предоставления заявителем в Росздравнадзор заявления и документов, перечисленных ниже.
Необходимо подчеркнуть, что ускоренная процедура регистрации предусматривает возможность регистрации изделий только с низкой степенью потенциального риска применения и предоставляется однократно в отношении одного наименования медицинского изделия одного производителя (изготовителя).
Для регистрации заявитель предоставляет:
- заявление о государственной регистрации;
- копию документа, подтверждающего полномочия уполномоченного представителя производителя (изготовителя);
- техническую документацию производителя (изготовителя) на медицинское изделие;
- эксплуатационную документацию производителя (изготовителя) на медицинское изделие, в том числе инструкцию по применению или руководство по эксплуатации медицинского изделия;
- фотографические изображения общего вида медицинского изделия вместе с принадлежностями, необходимыми для применения медицинского изделия по назначению (размером не менее 18 24 см);
- опись документов.
В случае недостаточности или некорректности предоставленных сведений на адрес заявителя направляется уведомление о необходимости устранения выявленных нарушений и (или) предоставления недостающих документов (материалов). Срок предоставления недостающих документов (материалов) составляет 5 рабочих дней.
С целью недопущения ошибок в документах регистрационного досье, поданного с целью государственной регистрации по Постановлению Правительства № 299, необходимо обратить внимание на следующее:
- Наименование медицинского изделия во всех документах регистрационного досье должно быть одинаковым.
- В случае, если организация, являющаяся местом производства, не является организацией, заявленной в качестве производителя, должен быть представлен документ о производственных отношениях между производителем и местом производства, содержащий в том числе сведения об ответственности за качество произведенной продукции, распределении ответственности перед третьими лицами.
- Содержание технической и эксплуатационной документации производителя (изготовителя) медицинского изделия должно соответствовать требованиям, утвержденным приказом Минздрава России от 19.01.2017 № 11н, в том числе в них должны быть указаны сведения о:
- потенциальных потребителях;
- применении в медицинской организации или вне ее;
- показаниях и противопоказаниях;
- хранении изделия;
- транспортировке изделия;
- утилизации;
- длительности использования изделия;
- гарантийных обязательствах и адресе для направления рекламаций;
- материалах/сырье, используемых для изготовления медицинского изделия;
- комплекте поставки.
Если в ходе рассмотрения досье у эксперта Росздравнадзора замечания к представленным документам отсутствуют, то готовится приказ о государственной регистрации медицинского изделия, включенного в Перечень.
В дальнейшем, в срок, не превышающий 150 рабочих дней со дня государственной регистрации, заявитель обязан представить в регистрирующий орган полный комплект документов, предусмотренный п. 57(10) Правил, в целях подтверждения государственной регистрации медицинского изделия:
- копию документа, подтверждающего полномочия уполномоченного представителя производителя (изготовителя);
- сведения о нормативной документации на медицинское изделие;
- документы, указанные в подпунктах «б», «в» и «г» пункта 57(2) Правил (в случае внесения в них изменений по результатам проведенных испытаний (исследований) медицинского изделия);
- документы, подтверждающие результаты технических испытаний медицинского изделия, выданные федеральным государственным бюджетным учреждением «Всероссийский научно-исследовательский и испытательный институт медицинской техники» Федеральной службы по надзору в сфере здравоохранения (далее – ФГБУ «ВНИИИМТ» Росздравнадзора);
- документы, подтверждающие результаты токсикологических исследований медицинского изделия, использование которого предполагает наличие контакта с организмом человека, выданные ФГБУ «ВНИИИМТ» Росздравнадзора;
- документы, подтверждающие результаты испытаний медицинского изделия в целях утверждения типа средств измерений (в отношении медицинских изделий, относящихся к средствам измерений, в сфере государственного регулирования обеспечения единства измерений, перечень которых утверждается Министерством здравоохранения Российской Федерации), выданные ФГБУ «ВНИИИМТ» Росздравнадзора;
- документы, подтверждающие результаты клинических испытаний медицинского изделия, проведенных в медицинской организации государственной системы здравоохранения, отвечающей требованиям, утвержденным Министерством здравоохранения Российской Федерации;
- копии документов, подтверждающих качество лекарственного препарата, фармацевтической субстанции, биологического материала и иного вещества, с использованием которых произведено медицинское изделие или которые входят в его состав и предназначены для применения только с учетом назначения медицинского изделия, определенного производителем, и выданных в соответствии с законодательством страны происхождения лекарственного препарата, фармацевтической субстанции, биологического материала и иного вещества;
- оригинал регистрационного удостоверения;
- опись документов.
Необходимо отметить, что документы, подтверждающие результаты технических испытаний, токсикологических исследований, испытаний в целях утверждения типа средств измерений согласно Постановлению Правительства № 299, должны быть выданы только ФГБУ «ВНИИИМТ» Росздравнадзора.
За период действия данного нормативного акта, ФГБУ «ВНИИИМТ» Росздравнадзора всего было проведено более 300 испытаний и исследований в целях подтверждения государственной регистрации.
После рассмотрения представленного пакета документов, согласно п. 57(10), экспертами Росздравнадзора он направляется в подведомственную экспертную организацию для проведения экспертизы качества, эффективности и безопасности медицинского изделия, аналогичной экспертизе, проводимой при стандартной процедуре государственной регистрации.
Решение о подтверждении государственной регистрации медицинского изделия принимается на основании положительного заключения от экспертного учреждения.
В случае получения отрицательного экспертного заключения, государственная регистрация медицинского изделия с низкой степенью потенциального риска применения отменяется.
Если в 150-дневный срок заявление о подтверждении государственной регистрации в Росздравнадзор не поступило, регистрационные удостоверения также подлежат отмене.
Необходимо отметить, что внесение изменений в документы, содержащиеся в регистрационном досье, а также получение дубликата регистрационного удостоверения невозможно до подтверждения регистрации.
Всего по Постановлению № 299 в 2020 г. выдано 1524 регистрационных удостоверения, из них основная доля приходится на отечественного производителя – 1353; остальные 171 – на зарубежного (рис. 1).
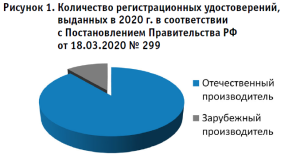
В настоящее время (по состоянию на 31.05.2021 г.) выдано 399 регистрационных удостоверений, из них:
- 351 – отечественного производителя
- 48 – зарубежного производителя.
Для облегчения подготовки документов с целью регистрации медицинских изделий, входящих в Перечень, заявителю рекомендуется пользоваться Методическими рекомендациями, размещенными на официальном сайте Росздравнадзора www.roszdravnadzor.gov.ru в разделе «Новости».
Упрощенная схема государственной регистрации серии (партии) медицинского изделия
Далее будет рассмотрена упрощенная схема государственной регистрации серии (партии) медицинского изделия, в соответствии с особенностями обращения медицинских изделий, в том числе государственной регистрации согласно перечню, утвержденному Постановлением Правительства Российской Федерации от 03.04.2020 № 430 «Об особенностях обращения медицинских изделий, в том числе государственной регистрации серии (партии) медицинского изделия» (далее – Перечень).
В данный Перечень входят аппараты искусственной вентиляции легких, изделия для диагностики in vitro, позволяющие выявлять новую коронавирусную инфекцию, а также средства индивидуальной защиты и др. Всего в Перечне содержится 363 вида медицинских изделий.
Обращаем внимание, что с целью регистрации серии (партии) медицинского изделия, заявление и документы, перечисленные ниже, направляются непосредственно в подведомственные экспертные организации, а не в Росздравнадзор:
- копия документа, подтверждающего полномочия уполномоченного представителя производителя (изготовителя) (при наличии);
- документы, подтверждающие принадлежность серии (партии) медицинского изделия заявителю на законных основаниях;
- техническая документация производителя (изготовителя) на медицинское изделие (при наличии);
- эксплуатационная документация производителя (изготовителя) на медицинское изделие, соответствующая требованиям, утвержденным Министерством здравоохранения Российской Федерации;
- фотографические изображения общего вида медицинского изделия вместе с принадлежностями, необходимыми для применения медицинского изделия по назначению (размером не менее 18 24 см);
- документы, подтверждающие результаты технических испытаний медицинского изделия, токсикологических исследований медицинского изделия, использование которого предполагает наличие контакта с организмом человека, клинических испытаний медицинского изделия, проведенных в соответствии с типовой программой испытаний в зависимости от вида медицинского изделия, разработанной экспертным учреждением (далее – Типовая программа) и опубликованной на официальных сайтах экспертных учреждений в информационно-телекоммуникационной сети «Интернет» (по применимости);
- иные документы, характеризующие качество, эффективность и безопасность медицинского изделия (при наличии);
- опись документов.
Следует обратить внимание на документ, подтверждающий принадлежность серии (партии) медицинского изделия заявителю на законных основаниях.
Отечественный производитель, являющийся также заявителем, может представить паспорт с указанием серии (партии) медицинского изделия.
Зарубежный производитель, являющийся также заявителем, может представить контракт, договор поставки, а также приложение к нему или инвойс, в котором четко идентифицированы номера серии (партии) изделия.
Необходимо отметить, что номера серий (партий) должны быть прописаны также в протоколах испытаний и исследований, представлены на фотографических изображениях.
Также для однозначной идентификации уполномоченного представителя производителя, должна быть представлена доверенность, в которой четко будут прописаны ИНН и ОГРН уполномоченной организации.
Технические испытания медицинского изделия, токсикологические исследования медицинского изделия, клинические испытания медицинского изделия проводятся в соответствии с Типовой программой испытаний в зависимости от вида медицинского изделия, которая разработана экспертным учреждением и опубликована на официальных сайтах экспертных учреждений.
В случае проведения в Российской Федерации испытаний (исследований) по программе, отличной от Типовой, экспертным учреждением определяется достаточность таких исследований (испытаний) для целей государственной регистрации серии (партии) медицинских изделий.
Необходимо иметь в виду, что требования, установленные Министерством здравоохранения Российской Федерации к проведению оценки соответствия медицинских изделий в форме технических испытаний, токсикологических исследований, клинических испытаний в целях государственной регистрации медицинских изделий, утвержденные приказом Минздрава России от 09.01.2014 № 2н, не являются обязательными для испытаний (исследований) медицинских изделий, включенных в Перечень.
Данное положение применяется до ликвидации угрозы возникновения чрезвычайной ситуации или ликвидации чрезвычайной ситуации. Медицинские изделия, не прошедшие указанную оценку соответствия, подлежат повторной регистрации в соответствии с законодательством Российской Федерации.
Таким образом, при регистрации серии (партии) медицинского изделия, проведение испытаний и исследований медицинского изделия является обязательным; испытания должны быть проведены, например, по Типовой программе.
Важно отметить, что внесение изменений допускается в наименование медицинского изделия в части изменения сведений о его заводском номере, номере серии при условии неизменности иных сведений, содержащихся в документах регистрационного досье, только для аппаратов искусственной вентиляции легких.
Регистрационное удостоверение на серию (партию) медицинского изделия выдается со сроком действия до 1 января 2022 г.
Реализация медицинских изделий с истекшим сроком действия регистрационного удостоверения не допускается, однако применение медицинского изделия возможно до окончания срока его годности.
Всего в 2020 г., согласно Постановлению Правительства Российской Федерации № 430, выдано 333 регистрационных удостоверения, из них:
- 66 – отечественного производителя;
- 267 – зарубежного производителя (рис. 2).

По состоянию на 31.05.2021 выдано 157 регистрационных удостоверений, из них:
- 33 – отечественного производителя;
- 124 – зарубежного производителя.
Организации, подведомственные Росздравнадзору, осуществляют консультирование по процедурам государственной регистрации медицинских изделий, предназначенных для диагностики новой коронавирусной инфекции, на безвозмездной основе.
Подводя итог, необходимо отметить, что Росздравнадзор в период пандемии смог в кратчайшие сроки допустить к обращению медицинские изделия, необходимые для борьбы с новой инфекцией и оснастить медицинские организации Российской Федерации необходимыми медицинскими изделиями.