LEGAL REGULATION OF THE CIRCULATION OF MEDICAL DEVICES AND MEDICINES
Circulation of medical devices: modern challenges
Author information
1 — Federal Service for Surveillance in Healthcare, 4, bld. 1, Slavyanskaya Square, Moscow, 109074, Russian Federation.
ORCID: https://orcid.org/0000–0002–9142–8808
2 — Federal Service for Surveillance in Healthcare, 4, bld. 1, Slavyanskaya Square, Moscow, 109074, Russian Federation.
3 — Federal State Budget Institution “Russian Scientific and Research Institute for Medical Engineering” of Roszdravnadzor, 24, bld. 16, Kashirskoye highway, Moscow, 115478, Russian Federation.
Published: 02.10.2023
Regulation of the circulation of medical devices is one of the main functions of the regulatory authorities of the healthcare system. The article presents the features of registration of medical devices that are relevant at the moment, describes the work of services created to support manufacturers on the issues of bringing medical devices to the market; the principles of a modern approach to monitoring the safety of medical devices on the territory of the Russian Federation are described. The features of inspection within the framework of national legislation are given when assessing the quality management system for the production of medical devices, depending on the potential risk of their use. Particular attention is paid to the importance of organizing proper maintenance and repair of medical equipment. Approaches leading to timely provision of the Russian market with safe and effective medical products have been formulated.
Keywords: quality and safety control of medical activities, tools to reduce risks in the provision of medical care, digitalization and informatization of healthcare, an index of citizens' satisfaction, principles of improving the quality and safety of medical activities
Background.
Рынок медицинских изделий в Российской Федерации является одним из быстро растущих и развивающихся в Евразийском экономическом союзе. Безопасное обращение медицинских изделий является неотъемлемой составной частью оказания медицинской помощи надлежащего качества. Качество, эффективность и безопасность применения медицинских изделий в медицинской организации напрямую влияет на безопасность медицинской деятельности.
Медицинская организация вправе использовать только зарегистрированные медицинские изделия, а также обеспечивает их надлежащее хранение, транспортировку, монтаж, наладку, применение, эксплуатацию, в том числе техническое обслуживание. Учитывая специфичность обеспечения безопасности обращения медицинских изделий, это направление деятельности является самостоятельным предметом контроля со стороны Росздравнадзора. Федеральная служба по надзору в сфере здравоохранения (далее – Росздравнадзор), являясь федеральным органом исполнительной власти, осуществляющим функции по контролю и надзору в сфере здравоохранения, реализует непрерывный мониторинг рынка медицинских изделий, уделяя в равной мере внимание вопросам регистрации медицинских изделий, безопасности обращения зарегистрированных медицинских изделий, инспектирования производства и ряду других.
Особенности регистрации медицинских изделий
Одним из ключевых этапов является регистрация медицинского изделия. В соответствии с п. 4 ст. 38 Федерального закона от 21.11.2011 № 323-ФЗ (в редакции от 11.06.2022, с изменениями от 13.07.2022) «Об основах охраны здоровья граждан в Российской Федерации» на территории России разрешается обращение медицинских изделий, которые прошли государственную регистрацию в порядке, установленном Правительством Российской Федерации, и медицинских изделий, прошедших регистрацию согласно международным договорам и актам, составляющим право Евразийского экономического союза (далее – ЕАЭС, Союз).
На официальном сайте Росздравнадзора в государственном реестре медицинских изделий и организаций (индивидуальных предпринимателей), осуществляющих производство и изготовление медицинских изделий, размещены сведения о 34 903 действующих регистрационных удостоверениях (РУ). Из них 13 772 (39%) РУ принадлежат отечественным производителям и 21 131 (61%) – зарубежным.
В соответствии с законодательством ЕАЭС в едином реестре медицинских изделий, зарегистрированных в рамках ЕАЭС, содержатся сведения о 33 зарегистрированных медицинских изделиях. Следует отметить, что большинство производителей – из Российской Федерации (17 изделий), 10 – из Республики Беларусь1.
Начиная с 2020 года система отечественного здравоохранения сталкивается с чрезвычайными ситуациями, требующими оперативного реагирования для решения наиболее острых проблем. Так, в 2020 году в связи со вспышкой новой коронавирусной инфекции, вызванной вирусом SARS-CoV-2 и чрезвычайными темпами ее распространения, был спровоцирован небывалый потребительский спрос на отдельные позиции медицинских изделий и, как следствие, последующий дефицит данных изделий. Возникла проблема выявления и диагностики вируса при отсутствии необходимых медицинских изделий на рынке, а существующая процедура государственной регистрации медицинских изделий, в соответствии с Правилами государственной регистрации медицинских изделий, утвержденными постановлением Правительства Российской Федерации от 27.12.2012 № 1416 «Об утверждении Правил государственной регистрации медицинских изделий» (далее – Постановление № 1416), оказалась недостаточно быстрой и гибкой для решения сложившейся ситуации.
Так, были введены две упрощенные процедуры регистрации в соответствии с постановлением Правительства Российской Федерации от 18.03.2020 № 299 «О внесении изменений в Правила государственной регистрации медицинских изделий» (далее – Постановление № 299) и постановлением Правительства Российской Федерации от 03.04.2020 № 430 «Об особенностях обращения медицинских изделий, в том числе государственной регистрации серии (партии) медицинского изделия» (далее – Постановление № 430).
Отличительной особенностью этих процедур стали скорость получения регистрационного удостоверения и упрощение процедуры – уведомительная регистрация для изделий, включенных в перечень.
Перечень медицинских изделий, предназначенных для использования в условиях военных действий, чрезвычайных ситуаций, предупреждения чрезвычайных ситуаций, профилактики и лечения заболеваний, представляющих опасность для окружающих, заболеваний и поражений, полученных в результате воздействия неблагоприятных химических, биологических, радиационных факторов, утвержденный Постановлением № 430, содержит уже 449 видов медицинских изделий вне зависимости от их класса потенциального риска применения.
Правительством Российской Федерации утверждено постановление от 01.04.2022 № 552 «Особенности обращения, включая особенности государственной регистрации, медицинских изделий в случае их дефектуры или риска возникновения дефектуры в связи с введением в отношении Российской Федерации ограничительных мер экономического характера» (с изменениями от 19.09.2022). Документ содержит два вида особенностей государственной регистрации медицинских изделий, а также вносит изменения в правила, определенные Постановлением № 1416, в части установления для всех медицинских изделий одноэтапной процедуры регистрации. Таким образом, экспертиза теперь проходит в один этап с одновременным анализом заявления о государственной регистрации, документов, оценкой полноты и результатов проведенных технических испытаний, токсикологических исследований, клинических испытаний, а также испытаний в целях утверждения типа средств измерений (при необходимости). Исключение составляют медицинские изделия, для которых необходимо проводить клинические испытания с участием человека.
Помимо ускорения и упрощения процедуры регистрации для медицинских изделий отечественного производства, включенных в перечень, за исключением имплантируемых и выпускаемых в стерильном виде медицинских изделий, допускается предоставление технических испытаний и токсикологических исследований, проведенных производителем или иной организацией, при условии возможности оценки применяемых методов и методик, а также перечня используемого оборудования.
Для медицинских изделий иностранного производства, за исключением программного обеспечения, которое является медицинским изделием, и медицинских изделий для диагностики in vitro, также предусмотрены послабления: возможность предоставлять копии документов, подтверждающих факт регистрации медицинского изделия в установленном порядке в стране-производителе, и документы, доказывающие клиническую эффективность и безопасность регистрируемого медицинского изделия.
При этом для медицинского изделия иностранного производства не предоставляются результаты клинических испытаний, проведенных на территории Российской Федерации в соответствии с порядком, установленным приказом Минздрава России от 30 августа 2021 года № 885н «Об утверждении Порядка проведения оценки соответствия медицинских изделий в форме технических испытаний, токсикологических исследований, клинических испытаний в целях государственной регистрации медицинских изделий». Для медицинских изделий с низкой степенью потенциального риска их применения (за исключением медицинских изделий, выпускаемых в стерильном виде), включенных в перечень медицинских изделий, решение о государственной регистрации принимается всего пять дней со дня получения заявления и необходимых документов. Однако, после получения регистрационного удостоверения заявитель обязан в соответствии с заключенным договором представить в ФГБУ «ВНИИИМТ» Росздравнадзора образец медицинского изделия для проведения технических испытаний, токсикологических исследований, испытаний в целях утверждения типа средств измерений (при необходимости).
Необходимо также сказать о постановлении Правительства Российской Федерации от 24 ноября 2021 года № 2026 «О незарегистрированных медицинских изделиях для диагностики in vitro», которое в период своего действия с 1 марта 2022 и до 1 марта 2028 г. определяет порядок предоставления, переоформления, подтверждения и отмены разрешения на применение медицинских изделий, которые предназначены для диагностики заболеваний путем проведения исследований образцов биологического материала человека вне его организма, изготовлены в медицинской организации и применяются в медицинской организации, их изготовившей.
Наряду с процедурой национальной регистрации установлен порядок:
- проведения процедур регистрации и экспертизы безопасности, качества и эффективности медицинских изделий;
- согласования экспертного заключения;
- урегулирования разногласий в отношении согласования экспертного заключения;
- внесения изменений в регистрационное досье (в том числе в уведомительном порядке);
- выдачи дубликата регистрационного удостоверения медицинского изделия;
- согласования экспертного заключения на зарегистрированное медицинское изделие;
- приостановления и (или) отмены действия (аннулирования) регистрационного удостоверения медицинского изделия на территории Союза в соответствии с п. 2 ст. 31 Договора о ЕАЭС от 29.05.2014 и п. 2 ст. 4 Соглашения от 23.12.2014 «О единых принципах и правилах обращения медицинских изделий (изделий медицинского назначения и медицинской техники) в рамках Евразийского экономического союза.
Минздрав России совместно с Росздравнадзором при проработке предложений, направленных на ускорение вывода на рынок востребованных медицинских изделий в условиях санкционного давления, выступили с инициативой проведения «государственного» консультирования, заключающегося не только в помощи производителям по отдельным вопросам регистрации, но и в проведении анализа всего регистрационного досье и помощи в его подготовке.
Данная инициатива нашла отражение в принятом правительством Российской Федерации в начале апреля 2022 г. Постановлении № 552, положениями которого подведомственные Росздравнадзору учреждения были наделены полномочиями по проведению такого консультирования.
Для деятельности Росздравнадзора произошла смена парадигмы с жесткого контроля на систему профилактики, предотвращение нарушений, а также на оптимизацию процедуры государственной регистрации медицинских изделий. Переориентация разрешительной деятельности на увеличение доверия к заявителям и контрольной деятельности – на предупреждение нарушений, стала подготовкой к выполнению новых задач, поставленных перед службой. Указанные особенности регистрации медицинских изделий значительно сокращают срок процедуры регистрации.
Сервисы для поддержки производителей
Подведомственный Росздравнадзору институт, ФГБУ «ВНИИИМТ» Росздравнадзора, в рамках поддержки производителей медицинских изделий в подготовке регистрационного досье для процедуры регистрации медицинских изделий запустил комплексную услугу поддержки производителей по выводу на рынок медицинских изделий, в состав которой входит:
- доработка/разработка технической/эксплуатационной документации (технические условия/выписка из технического файла, эксплуатационная документация производителя, сведения о нормативной документации, файл менеджмента риска);
- организация и проведение технических испытаний;
- организация и проведение токсикологических исследований;
- организация и проведение клинико-диагностических исследований in vitro;
- организация и проведение испытаний в целях утверждения типа средств измерений;
- организация и сопровождение клинических испытаний;
- предварительный анализ и оценка регистрационного досье.
Комплексная услуга позволяет выстроить с производителями медицинских изделий долгосрочные взаимоотношения:
- заключение рамочного договора на длительный срок сотрудничества;
- оказание необходимых услуг в формате «одно окно» в случае необходимости взаимодействия с другими испытательными центрами и лабораториями;
- индивидуальный подход к каждому заявителю.
За время реализации комплексной услуги ФГБУ «ВНИИИМТ» Росздравнадзора успешно завершило более 80 проектов с последующим получением регистрационных удостоверений на медицинские изделия. Средний срок с момента заключения договора до получения РУ составил 58 рабочих дней:
- для изделий класса риска 1 – 30 рабочих дней;
- для изделий класса риска 2а – 73 рабочих дня;
- для изделий класса риска 2б – 64 рабочих дня;
- для изделий класса риска 3 – 68 рабочих дней.
Средний срок оказываемых в рамках комплексной услуги работ составил:
- разработка комплекта технической и эксплуатационной документации – 31 рабочий день (в 3 раза быстрее, чем в 2021 году);
- проведение технических испытаний – 26 дней (в 3,5 раза быстрее, чем в 2021 году);
- проведение токсикологических исследований – 28 дней (в 4 раза быстрее, чем в 2021 году);
- предварительный анализ и оценка регистрационного досье – 10 дней (услуга появилась с момента вступления в силу Постановления № 552).
Мониторинг безопасности медицинских изделий
Принципы современного подхода к мониторингу безопасности медицинских изделий в Российской Федерации применяются с 2012 г., когда были введены Правила мониторинга безопасности медицинских изделий и Порядок сообщений субъектами обращения медицинских изделий обо всех случаях выявления побочных действий, не указанных в инструкции по применению или руководстве по эксплуатации медицинского изделия, о нежелательных реакциях при его применении, об особенностях взаимодействия медицинских изделий между собой, о фактах и об обстоятельствах, создающих угрозу жизни и здоровью граждан и медицинских работников при применении и эксплуатации медицинских изделий.
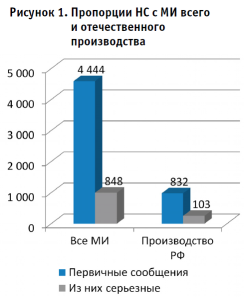
За период с 1 января 2019 г. по август 2023 г. всего в АИС Росздравнадзора зарегистрировано 5868 сообщений о неблагоприятных событиях, из них 4444 (76%) – первичные. Из числа первичных сообщений 848 (19%) – серьезные (смерть, угроза жизни, угроза здоровью и жизни).
Сравнительные данные о количестве первичных сообщений о неблагоприятных событиях (НС), связанных с отечественными изделиями, представлены на рисунке 1. Доля НС с изделиями производства РФ незначительна – 18,7% (832), из них доля серьезных событий – 12% (103), что ниже общего показателя. На рисунке 2 представлена динамика поступления сообщений от медицинских и фармацевтических работников и от производителей и их уполномоченных представителей.
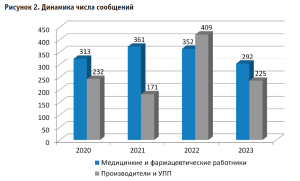
Таким образом, наблюдается рост количества сообщений от производителей, связанный с обязанностью учреждений здравоохранения сообщать производителю о неблагоприятных событиях, но, в целом, по-прежнему доля сообщений от медицинских организаций остается стабильной и превышает количество сообщений от производителей. Необходимо проведение разъяснительной работы с производителями и медицинским сообществом о необходимости сообщения об инцидентах с медицинскими изделиями.
Обеспечение качества при производстве МИ
Производство медицинских изделий является важнейшим этапом в обращении медицинских изделий. Сегодня законодательно внедряется инспектирование производства медицинских изделий как ключевой инструмент, позволяющий производителям использовать достоинства системы менеджмента качества (СМК) в качестве конкурентного преимущества современной концепции регулирования рынка МИ.
Внедрение предприятиями, изготавливающими медицинские изделия, сертифицированной по международным стандартам системы менеджмента качества имеет большую ценность. В частности, СМК позволит улучшить управляемость внутри предприятия, повысить конкурентоспособность, а главное – качество выпускаемой продукции. В глобальном масштабе это даст возможность обеспечить своевременный допуск качественных и безопасных МИ в государственные и частные медицинские организации, а также минимизировать риски причинения вреда жизни и здоровью пациентов.
Для внедрения системы менеджмента качества медицинских изделий производитель обязан разработать документированные требования к управлению рисками на всех этапах жизненного цикла медицинских изделий; определить процессы, необходимые для результативного функционирования СМК (далее – процессы), и должное осуществление данных процессов; определить последовательность и взаимосвязь процессов; определить критерии и методы, необходимые для обеспечения результативности как при осуществлении процессов, так и при управлении процессами; обеспечивать наличие условий производства, ресурсов и информации, необходимых для поддержания процессов и осуществления мониторинга процессов; осуществлять мониторинг, измерение (где применимо) и анализ процессов; принимать меры, необходимые для достижения запланированных результатов.
Способом оценки системы менеджмента качества производства медицинских изделий является проведение первичного, периодического (планового) и внепланового инспектирования производства. До 1 сентября 2022 года такой инструмент, как инспектирование производства, был доступен только при регистрации медицинских изделий в рамках Евразийского экономического союза. Основным документом является Решение Совета Евразийской экономической комиссии от 10 ноября 2017 г. № 106 «О Требованиях к внедрению, поддержанию и оценке системы менеджмента качества медицинских изделий в зависимости от потенциального риска их применения».
С 1 сентября 2022 года в действие вступило два постановления Правительства Российской Федерации от 09 февраля 2022 г. № 135 «Об утверждении Правил организации и проведения инспектирования производства медицинских изделий на соответствие требованиям к внедрению, поддержанию и оценке системы управления качеством медицинских изделий в зависимости от потенциального риска их применения» и № 136 «Об утверждении требований к внедрению, поддержанию и оценке системы управления качеством медицинских изделий в зависимости от потенциального риска их применения», в одно из которых уже были внесены изменения (постановление Правительства РФ от 29 декабря 2022 г. № 2517 «О внесении изменений в постановление Правительства Российской Федерации от 9 февраля 2022 г. № 135»).
Представленные постановления утверждают требования к внедрению, поддержанию и оценке системы управления качеством медицинских изделий в зависимости от потенциального риска их применения и регламентируют правила организации и проведения инспектирования производства медицинских изделий на соответствие требованиям к внедрению, поддержанию и оценке системы управления качеством медицинских изделий в зависимости от потенциального риска их применения в рамках регистрации медицинских изделий по национальным правилам, а именно правилам государственной регистрации медицинских изделий, утвержденным постановлением Правительства Российской Федерации от 27 декабря 2012 г. № 1416.
2023 год является переходным периодом, в который инспектирование производства в рамках национального законодательства осуществляется в добровольном порядке по инициативе производителя медицинского изделия или уполномоченного представителя производителя (изготовителя) медицинского изделия. Для производителей медицинских изделий класса потенциального риска применения 2а (для медицинских изделий, выпускаемых в стерильном виде), 2б или 3, которые хотят отложить проведение инспекции производства на 2024 год, при подаче документов на регистрацию или в целях внесения изменений в документы регистрационного досье необходимо в составе регистрационного досье медицинского изделия представить документы, подтверждающие наличие условий производства, а главное – сертификатов соответствия системы менеджмента качества требованиям стандарта ГОСТ ISO 13485-2017 или соответствующего международного стандарта ISO 13485, а также копии отчетов о ранее проведенных аудитах на соответствие указанным стандартам.
Отличительной особенностью инспектирования производства в рамках национального законодательства от законодательства ЕАЭС является то, что оценке системы управления качеством производителя медицинского изделия на соответствие требованиям к внедрению, поддержанию и оценке системы управления качеством медицинских изделий подлежат производители имплантируемых медицинских изделий, которые изготовлены по индивидуальным заказам пациентов, к которым предъявляются специальные требования по назначению медицинских работников. Производство индивидуальных изделий отличается от серийного производства подходами, уровнем контроля продукции, технологиями, например, использованием 3D-печати. Инспектирование производства набирает обороты. Первое инспектирование производства индивидуальных имплантатов было проведено в середине августа 2023 г. совместными силами сотрудников ФГБУ «ВНИИИМТ» Росздравнадзора и Федеральной службы по надзору в сфере здравоохранения.
Надлежащее техническое обслуживание и ремонт медицинской техники
Техническое обслуживание медицинской техники в гарантийный и послегарантийный период является обязательным условием ее безопасной эксплуатации и эффективного применения по назначению. Уход с рынка России западных производителей, их сервисных и логистических служб, принудительное нарушение цепочек поставок запасных частей и комплектующих создают угрозу работоспособности медицинского оборудования, а, следовательно, ограничивают доступность медицинских услуг для населения. МИ многих производителей могут полноценно обслуживаться лишь специально подготовленными производителем техническими специалистами. Также многие производители МИ не предоставляют сторонним сервисным службам документацию, ключи и пароли доступа. Сегодня в большинстве регионов Российской Федерации существует децентрализованная модель технического обслуживания и ремонта. Все проблемы приходится решать самостоятельно руководителям медицинских организаций. Кроме того, низкий общий уровень подготовки технических заданий государственных контрактов на обслуживание медицинских изделий и существующая система государственных закупок могут приводить к существенным срокам простоя МИ в периоды ремонта, что, как следствие, ставит под угрозу жизнь и здоровье граждан, а это недопустимо.
В 2023 г. на базе ФГБУ «ВНИИИМТ» Росздравнадзора был создан единый центр компетенций по сервисному обслуживанию медицинской техники, основной задачей которого является повышение эффективности функционирования системы здравоохранения путем создания надежного механизма взаимодействия всех участников сферы обращения медицинских изделий – органов государственной власти субъектов Российской Федерации, медицинских организаций, а также организаций, осуществляющих техническое обслуживание и ремонт медицинской техники, для достижения оптимальных решений при эксплуатации медицинских изделий в медицинских организациях страны.
В зоне особого внимания находятся медицинские организации новых регионов: Донецкой Народной Республики, Луганской Народной Республики, Запорожской и Херсонской областей, оборудование которых длительное время не обслуживалось и находится в нерабочем состоянии. Наша общая задача – обеспечить бесперебойную работу и оказание медицинской помощи гражданам с использованием современных, безопасных, работоспособных медицинских изделий.
Выводы
Современные вызовы, стоящие перед нашей страной, обязывают нас к четким, быстрым и эффективным решениям и действиям для обеспечения слаженной работы всей системы здравоохранения. Формирование нормативного правового регулирования, соблюдение действующего законодательства, превентивные профилактические мероприятия, необходимый контроль и надзор позволит обеспечить российский рынок безопасными и эффективными медицинскими изделиями, что в конечном итоге повысит доступность, своевременность и качество медицинской помощи для граждан Российской Федерации.
_______________________________________________________________________
1 URL: http://government.ru/docs/all/138877/