КОНТРОЛЬ КАЧЕСТВА ЛЕКАРСТВЕННЫХ СРЕДСТВ
Сравнительный анализ методов количественного определения полисорбата-20 в лекарственных препаратах на основе рекомбинантных белков
Информация об авторах
1 — Федеральное государственное бюджетное учреждение «Информационно-методический центр по экспертизе, учету и анализу обращения средств медицинского применения» Федеральной службы по надзору в сфере здравоохранения, 109012, Российская Федерация, Москва, Славянская площадь, д. 4, стр. 1.
ORCID: https://orsid.org/0000-0002-9667-1014
2 — Федеральное государственное бюджетное учреждение «Информационно-методический центр по экспертизе, учету и анализу обращения средств медицинского применения» Федеральной службы по надзору в сфере здравоохранения, 109012, Российская Федерация, Москва, Славянская площадь, д. 4, стр. 1.
ORCID: https://orcid.org/0000-0001-5637-8836
Опубликовано: 04.10.2022
В работе проведено сравнение методов количественного определения полисорбата-20 в составе лекарственного препарата, представляющего собой моноклональное антитело. Для исследования был использован метод определения продуктов щелочного гидролиза с последующей дериватизацией и метод анализа флуоресценции мицелл (FMA). Основное внимание было уделено определению сходимости полученных результатов в случае использования одного метода, сравнению результатов, полученных различными методами на аналогичных препаратах или модельных смесях, а также оценке степени влияния активного вещества на определение полисорбата-20 в каждом из используемых методов.
Ключевые слова: полисорбат-20, моноклональные антитела, количественное определение
Введение
Лаборатории контроля качества лекарственных средств ФГБУ «Информационно-методический центр по экспертизе, учету и анализу обращения средств медицинского применения» («ИМЦЭУАОСМП») Росздравнадзора в рамках полномочий по государственному контролю (надзору) или вводу в гражданский оборот лекарственных препаратов проводят испытания большого спектра лекарственным средств, в том числе и препаратов моноклональных антител (ОФС.1.7.1.0014.18). Действующими веществами в данных препаратах являются белковые молекулы сложной структуры. При производстве таких лекарственных средств производитель сталкивается с тем, что белок имеет свойство агрегировать при хранении или механическом воздействии, или денатурировать при лиофилизации или транспортировке, а также имеет способность сорбироваться на стенках емкости, из-за чего снижается его биологическая активность. Для решения этой проблемы используются неионогенные поверхностно-активные вещества, способные стабилизировать молекулы белка в растворе.
Примером таких веществ являются полисорбаты – безопасный класс соединений, проявляющих свои свойства даже в крайне малых концентрациях. К числу таких соединений относится полисорбат-20. Его молекула состоит из гидрофобной части, представленной ядром сорбитана с полиоксиэтиленовыми цепями, а также гидрофильная часть – остаток лауриновой кислоты (рис. 1). Полисорбат-20 является наиболее гидрофильным соединением в ряду применяемых полисорбатов [1]. Полисорбаты мало сорбируются на поверхности белка и обладают незначительным противовирусным действием, что делает их эффективным стабилизатором для пептидных и белковых препаратов и особенно для препаратов, получаемых с использованием культур клеток. Роль стабилизатора крайне важна при производстве и хранении лекарственных средств, поэтому также важно контролировать их содержание [2, 8].
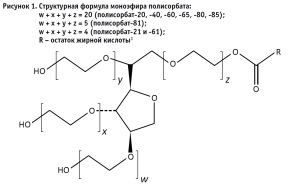
Государственная фармакопея Российской Федерации и Фармакопея Евразийского экономического союза не дают указаний на единый метод количественного определения полисорбатов и, в частности, полисорбата-20. Главными проблемами контроля соединений данной группы являются следующие:
- соединения не имеют хромофорных групп, то есть не могут быть определены прямым детектированием на спектральных детекторах;
- содержатся в низких концентрациях в лекарственном препарате, то есть требуют применения высокочувствительных методик;
- создают с молекулами белка комплекс, из которого сложно выделить исследуемое вещество.
По этим причинам невозможно использовать стандартные методы количественного определения. Для решения данной проблемы исследователями были предложены несколько подходов, позволяющих в той или иной степени разрешить указанные противоречия. Заявленные производителями методы определения можно условно разделить на две большие подгруппы: определение непосредственно полисорбата и определение продуктов его гидролиза [2, 3, 10].
Для определения интактного полисорбата чаще всего в нормативной документации встречаются методы обращенно-фазовой хроматографии с детектором заряженного аэрозоля (CAD), испарительным детектором по светорассеянию (ELSD), спектрофотометрическое определение после реакции с кобальта тиоционат аммонием и последующей экстракцией в органическую фазу [9, 10]. Эти методы занимают много времени, сложны в воспроизведении, а также, в случае с хроматографией, требуют наличия дорогостоящих узкопрофильных детекторов.
Еще одним способом количественного определения непосредственно полисорбата является проточно-инжекционный метод с анализом флуоресценции мицелл (FMA). Данный метод выполняется с помощью хроматографа, но не требует использования колонки для разделения, вместо этого используется реакционная петля, в которой происходит образование мицелл полисорбата с N-фенил-1-нафтиламином, входящим в состав подвижной фазы. Участвующий в реакции краситель практически не флуоресцирует в водной среде, но хорошо делает это в гидрофобной среде, в данном случае после того, как встраивается в гидрофобное ядро образовавшихся мицелл полисорбата [4, 5, 11, 12]. Анализ проводится с помощью флуориметрического детектора, не требует специфической пробоподготовки образца и занимает короткое время.
В случае с определением продуктов гидролиза полисорбата проводят анализ обращенно-фазовой хроматографией со спектрофотометрическим детектором образовавшихся жирных кислот, либо их дериватов. Данный подход требует длительной, многоступенчатой пробоподготовки. Дополнительной сложностью является то, что из-за особенностей синтеза, серии полисорбата могут иметь различия в своем составе, например, иметь отличающееся соотношение эфиров полиоксиэтиленсорбитана и различных жирных кислот, например, содержание лауриновой кислоты в полисорбате-20 может быть от 40 до 60% [13]. По этой причине в методах контроля с применением гидролиза необходимо в качестве стандарта использовать полисорбат той же серии, которая была задействована при производстве лекарственного препарата [6].
В данной работе проведено сравнение методов количественного определения полисорбата-20 в составе лекарственного препарата, представляющего собой моноклональное антитело. Для исследования были использованы метод определения продуктов щелочного гидролиза с последующей дериватизацией и метод анализа флуоресценции мицелл (FMA). Основное внимание было уделено определению сходимости полученных результатов в случае использования одного метода, сравнению результатов, полученных различными методами на аналогичных препаратах или модельных смесях, а также оценке степени влияния активного вещества на определение полисорбата-20 в каждом из используемых методов.
Материалы и методы
Объект исследования и сравнения. В качестве объекта исследования использовался лекарственный препарат, представляющий собой моноклональное антитело и содержащий в составе полисорбат-20 в качестве стабилизатора. В роли стандартного образца использовался полисорбат-20 производства PanReac (кат. № 142312), с содержанием воды 2,4%.
Оборудование и реактивы. Анализ методом обращенно-фазовой высокоэффективной жидкостной хроматографии с предварительным гидролизом и дериватизацией (Метод 1) проводился с использованием следующего оборудования: твердотельный термостат TDB-120, центрифуга MiniSpin Plus, хроматограф жидкостный высокоэффективный Ultimate 3000, оборудованный спектрофотометрическим детектором с диодной матрицей. Для разделения продуктов реакции была использована хроматографическая колонка YMC-Pack ODS-AQ, 150 4,6 мм, 3 мкм. Реактивы: ацетонитрил, фосфорная кислота концентрированная, калия гидроксид, муравьиная кислота, триэтиламин, 4-бромометил-7-метоксикумарин, N,N-диметилформамид.
Анализ проточно-инжекционным методом с измерением флуоресценции мицеллы (Метод 2) проводился с использованием следующего оборудования: хроматограф жидкостный высокоэффективный Agilent 1200, оборудованный флуориметрическим детектором и реакционной петлей Knitted reactor coil, 5м 0,5мм, объемом 9,8 мл. Реактивы: ацетонитрил, кислота хлористоводородная, натрия хлорид, Brij 35, трис(гидроксиметил)аминометан, N-фенил-1-нафтиламин.
Для приготовления раствора плацебо использовали следующие реактивы: L-гистидина моногидрохлорид моногидрат, L-гистидин, -трегалозы дигидрат. Все используемые реактивы имели квалификацию «химически чистые» (х.ч.) или «чистые для анализа» (ч.д.а.). Все используемое оборудование на момент проведения испытаний имело действующие сертификаты поверки.
Подготовка растворов. Раствор плацебо: 0,56 г L-гистидина моногидрохлорида моногидрата, 0,36 г L-гистидина, 22,7 г , -трегалозы дигидрата растворяли в 900 мл воды очищенной в стеклянном стакане, контролировали значение рН (рН = 6,01). Затем переносили полученный раствор в мерную колбу вместимостью 1000 мл и доводили водой очищенной до метки. Фильтровали полученный раствор через нейлоновый мембранный фильтр с диаметром пор 0,22 мкм. Раствор хранили при температуре от 2 до 8 °C (срок хранения – 6 месяцев).
Раствор 5М калия гидроксида для проведения гидролиза (Метод 1): около 5 мл воды очищенной помещали в химический стакан вместимостью 10 мл и небольшими порциями при постоянном перемешивании добавляли 2,8 г калия гидроксида, тщательно перемешивая до полного растворения навески. Полученный раствор количественно переносили в мерную колбу вместимостью 10 мл, после чего доводили объем раствора до метки водой очищенной и перемешивали. Раствор использовали свежеприготовленным.
Дериватизирующий реагент (Метод 1): к 50 мг триэтиламина прибавляли 1,25 мл ацетонитрила и сразу же тщательно перемешивали. К 30 мг 4-бромометил-7-метоксикумарина прибавляли 3,0 мл N,N-диметилформамида и тщательно перемешивали до полного растворения. Далее к 2,0 мл раствора 4-бромометил-7-метоксикумарина прибавляли 1,0 мл приготовленного раствора триэтиламина и перемешивали. Раствор использовали свежеприготовленным. 4% раствор муравьиной кислоты (Метод 1): 5,0 мл воды очищенной помещали в мерную колбу вместимостью 10 мл, прибавляли 400 мкл муравьиной кислоты, перемешивали, после чего доводили объем раствора до метки водой очищенной и еще раз перемешивали. Раствор использовали свежеприготовленным.
Раствор N-фенил-1-нафтиламина 10 мМ (Метод 2): в мерную колбу объемом 50 мл помещали 0,11 г N-фенил-1-нафтиламина, растворяли в ацетонитриле и доводили до метки этим же растворителем.
Раствор Brij 35 3% (Метод 2): 3,0 г Brij 35 помещали в мерную колбу объемом 100 мл, растворяли в 40 мл воды, после чего доводили до метки тем же растворителем. Подвижная фаза А (Метод 1): использовали ацетонитрил.
Подвижная фаза Б (Метод 1): около 100 мл воды очищенной помещали в мерную колбу вместимостью 500 мл, затем прибавляли 5,0 г фосфорной кислоты концентрированной, затем доводили объем раствора водой очищенной до метки, после чего перемешивали. Раствор использовали в течение 1 суток при хранении при температуре от 15 до 25 °С.
Подвижная фаза В (Метод 2): в мерную колбу объемом 2000 мл помещали 17,5 г натрия хлорида и 12,1 г трис(гидроксиметил)аминометана, растворяли данные навески в 1800 мл воды, после чего доводили значение рН раствора хлористоводородной кислотой концентрированной до рН = 8,0, далее добавляли в мерную колбу 100 мл ацетонитрила и доводили до метки водой. Раствор фильтровали через мембранный фильтр с диаметром пор 0,45 мкм, после чего к полученному раствору прибавляли 1 мл 10 мМ раствора N-фенил-1-нафтиламина и 1 мл 3% раствора Brij 35, тщательно перемешивали полученный раствор.
Подготовка стандартных и испытуемых образцов. Исходные растворы стандартных образцов: готовили два раствора – первый использовали для приготовления контрольного раствора стандартного образца, второй – для приготовления калибровочных стандартных растворов.
Раствор 1: 106,3 мг полисорбата-20 помещали в химический стакан вместимостью 25 мл, затем прибавляли около 10 мл воды очищенной и перемешивали с помощью магнитной мешалки до полного растворения навески. Раствор количественно переносили в мерную колбу вместимостью 100 мл, доводили объем раствора до метки водой очищенной и перемешивали. Контрольный раствор стандартного образца с концентрацией 0,1 мг/мл готовили в двух экземплярах – на водной основе и с раствором плацебо, в качестве растворителя. Для этого в мерную колбу вместимостью 25 мл помещали 2,410 мл Раствора 1, после чего доводили водой очищенной, либо раствором плацебо до метки. Растворы использовались свежеприготовленными.
Раствор 2: 102,2 мг полисорбата-20 помещали в химический стакан вместимостью 25 мл, затем прибавляли около 10 мл воды очищенной и перемешивали с помощью магнитной мешалки до полного растворения навески. Раствор количественно переносили в мерную колбу вместимостью 100 мл, доводили объем раствора до метки водой очищенной и перемешивали.
Далее готовили раствор полисорбата-20 с концентрацией 0,4 мг/мл в двух экземплярах – на водной основе и с раствором плацебо в качестве растворителя. Для этого в мерную колбу вместимостью 25 мл помещали 10,025 мл Раствора 2, после чего доводили водой очищенной либо раствором плацебо до метки. Из полученного раствора готовили калибровочные стандартные растворы в пластиковых пробирках вместимостью 1,5 мл, как указано в таблице 1.
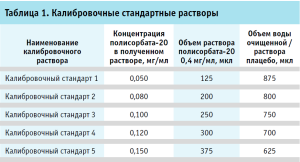
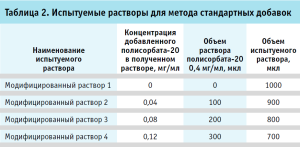
Испытуемые растворы. Во флакон с лиофилизатом препарата вносили 6,0 мл воды для инъекций. Теоретическая концентрация полисорбата-20 во флаконе составляла при этом 0,1 мг/мл. Восстановленный раствор использовали свеже приготовленным. Отдельно готовили испытуемые растворы для метода стандартных добавок. Для этого к испытуемому раствору добавляли раствор полисорбата-20 с концентрацией 0,4 мг/мл (табл. 2).
Подготовка испытуемых, контрольных и калибровочных растворов. Для Метода 1 гидролиз проводится в чистых пластиковых пробирках объемом 0,5 мл. 100 мкл 5 М раствора калия гидроксида смешивали с 100 мкл растворов образцов и инкубировали полученные растворы при температуре 80°С в течение 60 мин. Центрифугировали микропробирки с образцами при 9000 g в течение 1 мин для полного удаления конденсата с внутренней поверхности крышек и стенок микропробирок. Добавляли 50 мкл 5 М раствора калия гидроксида и 200 мкл ацетонитрила. Перемешивали с помощью вибромешалки в течение 5 минут для достижения полного перехода гидролизата полисорбата-20 (лауриновой кислоты) в ацетонитрил. Центрифугировали образцы при 9000 g в течение 1 мин.
В новые пластиковые пробирки помещали 100 мкл дериватизирующего реагента, после чего добавляли по 100 мкл верхнего слоя образцов (ацетонитрил). Перемешивали с помощью вибромешалки, затем центрифугировали при 9000 g в течение 1 мин.
Инкубировали полученные растворы при температуре 60°С в течение 60 мин, после чего центрифугировали при 9000 g в течение 1 мин.
Добавляли ко всем образцам 66 мкл 4% раствора муравьиной кислоты и перемешивали для нейтрализации среды проб после щелочного гидролиза.
Для Метода 2 испытуемый, контрольный и калибровочный растворы использовались без дополнительной пробоподготовки.
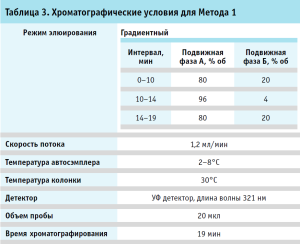
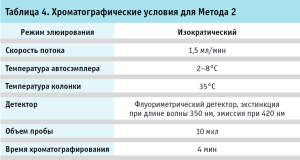
Хроматография. Хроматографические условия для Метода 1 и 2 приведены в таблицах 3, 4.
Результаты и обсуждение
Для Метода 1 в качестве пика полисорбата-20 принимали пик деривата лауриновой кислоты (4-лауроилоксиметил-7-метоксикумарин), время удерживания около 9,1 мин.
По полученным данным строили калибровочный график, откладывая по оси абсцисс содержание полисорбата-20 в разведениях калибровочных стандартных образцов (в мг/мл), а по оси ординат – площадь пика деривата лауриновой кислоты для Метода 1 и площадь пика полисорбата-20 для Метода 2 (в единицах подсчета площади) (рис. 2–5).
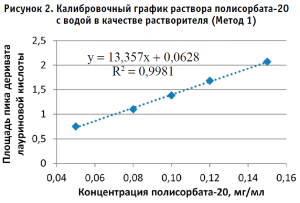
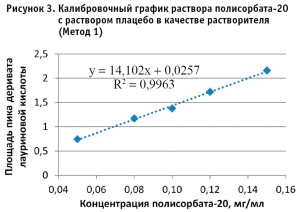
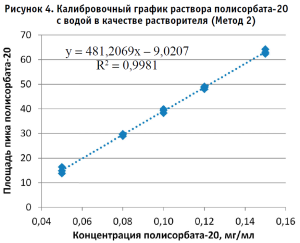
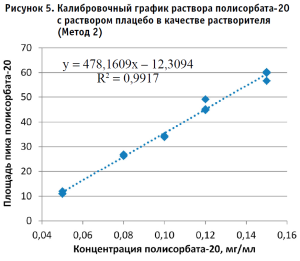
Методом наименьших квадратов рассчитывали уравнение, описывающее зависимость вида:
y = kx + b,
где:
y – это площадь аналита;
x – концентрация полисорбата-20 (мг/мл);
k – угол наклона;
b – отрезок, отсекаемый по оси Y.
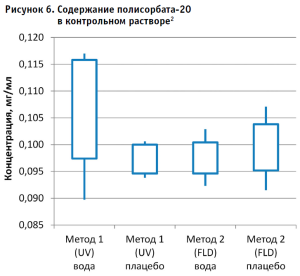
Далее рассчитывали содержание полисорбата-20 в контрольном растворе (рис. 6) по каждому из полученных уравнений, используя формулу:
Концентрация полисорбата-20 = (площадь пика аналита b) / k
Рассчитывали стандартное отклонение и доверительный интервал 95% (± 2 стандартных отклонения от среднего значения).
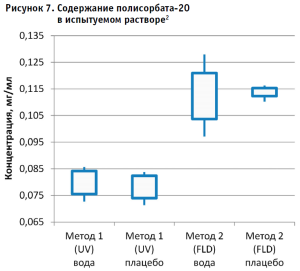
Затем аналогичным образом обрабатывали результаты для испытуемого образца, используя для расчета построенные калибровочные графики (рис. 2–5, 7). По результатам проведенных экспериментов можно сделать следующие заключения:
- Использование раствора плацебо без действующего вещества в качестве растворителя при приготовлении калибровочных и контрольных растворов не оказывает значительного влияния на полученные результаты, что позволяет пренебречь данным раствором и использовать вместо него воду очищенную для упрощения и ускорения проведения анализа. Следует отметить, что данные результаты относятся к использованию раствора плацебо указанного состава.
- Наиболее интересным результатом эксперимента следует признать тот факт, что результаты содержания полисорбата-20 в испытуемом образце, полученные разными методами, достоверно отличаются и не перекрывают доверительные интервалы 95% между собой. В то же время результаты контрольного раствора, не содержащего белка, находятся в одном диапазоне значений.
- Литературные данные свидетельствуют, что полисорбаты обладают невысокой, но ненулевой адсорбцией на белках, поэтому пренебрегать таким явлением не следует. Особенно это важно для лекарственных композиций, в которых белок составляет значительную долю состава. Различия в степени адсорбции также могут быть вызваны структурой целевого белка, в том числе его зарядом и другими физико-химическими характеристиками. Так как белок, находящийся в препарате, сорбирует на себе полисорбат, можно сделать заключение, что это может влиять на прохождение гидролиза и полноту извлечения лауриновой кислоты, в связи с чем могли быть получены различия в результатах.
После обсуждения было принято решение повторить эксперимент, прибегнув к методу стандартных добавок [7]. При обработке результатов, полученных при использовании метода стандартных добавок, обращает на себя внимание тот факт, что при применении Метода 2 (без дериватизации), полученные значения концентраций полисорбата-20 в испытуемых растворах с добавками соответствуют теоретическим ожиданиям. (табл. 5). В то же время практические значения, полученные Методом 1, для всех растворов с внесенными добавками были ниже теоретических, причем не имело значения, на воде очищенной или растворе плацебо был приготовлен испытуемый раствор. Для более точной оценки полученных результатов для Метода 1 был построен график зависимости в координатах аналитический сигнал – концентрация добавленного стандартного раствора полисорбата-20 (рис. 8) В соответствии с рекомендациями по использованию расчетов по методу стандартных добавок, отрезок, отсекаемый полученной прямой по оси Х, равен концентрации полисорбата-20 в растворе без добавок и составляет 0,10 мг/мл, что является значением, близким к теоретическому.
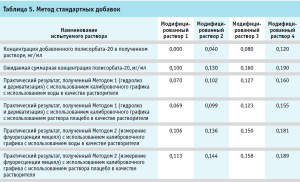
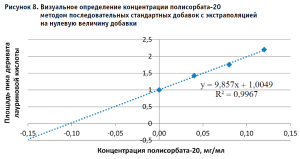
Такой результат позволяет предположить, что в исходном растворе испытуемого лекарственного средства существуют условия, исходно препятствующие полному гидролизу полисорбата-20, что соответствует имеющимся литературным данным о характере поведения аналитов, адсорбирующихся на белковых молекулах.
При этом показано, что использование метода стандартных добавок в данном случае позволяет решить возникающую проблему и нивелировать степень влияния на получаемые результаты систематической ошибки, возникающей вследствие частичной адсорбции полисорбата-20 на белке.
Заключение
В результате проведенных экспериментов было показано, что при сравнительном использовании метода определения продуктов щелочного гидролиза с последующей дериватизацией и метода анализа флуоресценции мицелл для определения концентрации полисорбата-20 в растворе лекарственного препарата, содержащем белковые молекулы, не удается добиться сходимых результатов в том случае, если предложенный дизайн метода дериватизации предполагает оценку результатов испытания только по одному вводу испытуемого раствора, что связано с возможным неполным гидролизом молекул полисорбата-20, адсорбированных на белковых молекулах. В то же время, использование данного метода в модификации метода стандартных добавок позволяет получить правильные результаты, что позволяет рекомендовать использование именно такого дизайна испытания для определения количественного содержания полисорбата-20 в растворах лекарственных средств, содержащих белковые молекулы.
Для уточнения данных рекомендаций авторами запланирована серия дальнейших экспериментов.
_______________________________________________________________________
1 например, лауриновой – в случае с полисорбатом-20.
2 Примечание: «усы» – 95% доверительного интервала, границы ящика – минимальное и максимальное значения полученных концентраций.
- Evaluation Report of Food Additives “Polysorbate 20, Polysorbate 60, Polysorbate 65 and Polysorbate 80”. Food Safety Commission. Office Location of the FSC: Japan. Tokyo. 2007.
- Дегтерев М.Б. Способ измерения количества полисорбата-80 с применением щелочного гидролиза образца с последующей ВЭЖХ. – 2018.
- Гроховский В.И. Определение полисорбатов в биотехнологических препаратах / В.И. Гроховский, А.А. Бендрышев, С.В. Швец, Д.А. Орлов, О.А. Ваганова // Разработка и регистрация лекарственных средств. – 2017. – № 2. – С. 160–165.
- Mahle H.C., Ravuri S.K.K. Pharmaceutical formulation for proteins. November 22. 2012. US Patent App. 13/574,071.
- Tomlinson A. et al. Polysorbate 20 degradation in biopharmaceutical formulations: quantification of free fatty acids, characterization of particulates, and insights into the degradation mechanism // Molecular pharmaceutics. 2015; Vol. 12 (11): 3805–3815.
- Dang H.V. Eccleston. Composition analysis of two batches of polysorbate 60 using MS and NMR techniques / H.V. Dang, A.I. Gray, D. Watson, C.D. Bates, P. Scholes, G.M. // Journal of Pharmaceutical and Biomedical Analysis. 2006; Vol. 40 (I.5): 1155–1165.
- Зенкевич И.Г., Морозова Т.Е. Особенности метода стандартной добавки для количественного определения аналитов в сложных матрицах, обладающих сорбционными свойствами // Аналитика и контроль. – 2010 – № 3. – С. 164–171.
- Вебер А. и др. Способ определения полисорбата. – 2016.
- Lippold S. et al. Impact of mono-and poly-ester fractions on polysorbate quantitation using mixed-mode HPLC-CAD/ELSD and the fluorescence micelle assay // Journal of Pharmaceutical and Biomedical Analysis. 2017; Vol. 132: 24–34.
- Аскретков А.Д. Определение полисорбатов спектрофотометрическим методом в лекарственных препаратах на основе рекомбинантных белков / А.Д. Аскретков, П.М. Исайкина, С.А. Черепушкин, Н.В. Орлова // Разработка и регистрация лекарственных средств. – 2017. – № 3. – С. 124–129.
- Khossravi M. et al. Analysis methods of polysorbate 20: A new method to assess the stability of polysorbate 20 and established methods that may overlook degraded polysorbate 20 // Pharmaceutical research. 2002; Vol. 19 (5): 634–639.
- Lapelosa M., Patapoff T. W., Zarraga I. E. Molecular simulations of micellar aggregation of polysorbate 20 ester fractions and their interaction with N-phenyl-1-naphthylamine dye //Biophysical chemistry. 2016; Vol. 213: 17–24.
- Rowe R. C., Sheskey P., Quinn M. Handbook of pharmaceutical excipients. – LibrosDigitales-Pharmaceutical Press, 2009.