РЕГУЛИРОВАНИЕ ОБРАЩЕНИЯ МЕДИЦИНСКИХ ИЗДЕЛИЙ
Специфика подготовки регистрационного досье в целях внесения изменений в регистрационные документы в связи с вступлением в силу постановления Правительства Российской Федерации от 01.04.2022 № 552
Информация об авторах
1 — Федеральная служба по надзору в сфере здравоохранения, 109074, Российская Федерация, Москва, Славянская площадь, д. 4, стр. 1.
Опубликовано: 30.09.2022
Статья посвящена вопросам подготовки регистрационного досье в целях внесения изменений в регистрационные документы в связи с вступлением в силу постановления Правительства Российской Федерации от 01.04.2022 № 552 «Об утверждении особенностей обращения, включая особенности государственной регистрации, медицинских изделий в случае их дефектуры или риска возникновения дефектуры в связи с введением в отношении Российской Федерации ограничительных мер экономического характера».
Ключевые слова: медицинские изделия, особенности обращения медицинских изделий в случае дефектуры, внесение изменений в регистрационное досье, особенности государственной регистрации медицинских изделий
Особенностями обращения (включая особенности государственной регистрации) медицинских изделий в случае их дефектуры или риска возникновения дефектуры в связи с введением в отношении Российской Федерации ограничительных мер экономического характера, утвержденными постановлением Правительства Российской Федерации от 01.04.2022 № 552 (далее соответственно – Особенности, Постановление), определены особенности обращения медицинских изделий, в том числе особенности государственной регистрации медицинских изделий, а также порядок внесения изменений в документы, содержащиеся в регистрационном досье, на:
- медицинские изделия, указанные в перечне медицинских изделий, подлежащих обращению в соответствии с Постановлением (далее – Перечень), формируемом в соответствии с п. 4 Особенностей;
- медицинские изделия с низкой степенью потенциального риска их применения (за исключением медицинских изделий, выпускаемых в стерильном виде), включенные в Перечень (далее – медицинские изделия с низкой степенью потенциального риска);
- медицинские изделия отечественного производства.
Порядок внесения изменений в документы, содержащиеся в регистрационном досье на медицинское изделие, включенное в Перечень, в соответствии с Особенностями и Правилами государственной регистрации медицинских изделий, утвержденных постановлением Правительства Российской Федерации от 27.12.2012 № 1416 «Об утверждении Правил государственной регистрации медицинских изделий» (далее – Правила), приведен в таблице 1.
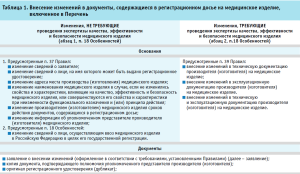

Особенности внесения изменений в документы, содержащиеся в регистрационном досье медицинского изделия с низкой степенью потенциального риска
Постановлением также утверждены Особенности государственной регистрации медицинских изделий с низкой степенью потенциального риска (пункты 23-29 Особенностей).
Вместе с тем, согласно п. 28 Особенностей, внесение изменений в документы, содержащиеся в регистрационном досье медицинского изделия с низкой степенью потенциального риска, осуществляется после принятия решения о государственной регистрации медицинского изделия в соответствии с Правилами.
Особенности внесения изменений в документы, содержащиеся в регистрационном досье на медицинское изделие отечественного производства
В соответствии с Особенностями, под медицинским изделием отечественного производства понимается медицинское изделие, в регистрационном удостоверении на которое, либо в заявлении о государственной регистрации которого в качестве производителя (изготовителя) указано юридическое лицо или физическое лицо, зарегистрированное в качестве индивидуального предпринимателя, являющееся резидентом Российской Федерации, а также имеющее место производства (производственную площадку) на территории Российской Федерации (далее – медицинское изделие отечественного производства). При этом регистрационное удостоверение должно быть выдано в соответствии с Особенностями или Правилами.
К изменениям, вносимым в документы, содержащиеся в регистрационном досье на медицинское изделие отечественного производства, зарегистрированное в соответствии с Особенностями или Правилами, требующими проведения экспертизы качества, эффективности и безопасности медицинского изделия, относятся:
- изменение сведений о покупных изделиях, сырье, материалах и комплектующих;
- изменение сведений о составных частях, запасных частях и принадлежностях.
Для внесения изменений в документы, содержащиеся в регистрационном досье, заявитель не позднее чем через 30 рабочих дней со дня изменения соответствующих данных, представляет в Росздравнадзор:
- заявление;
- копию документа, подтверждающего полномочия уполномоченного представителя производителя (изготовителя);
- документы и сведения о соответствующих изменениях, в том числе документы, подтверждающие изменения и свидетельствующие о том, что внесение этих изменений не влечет за собой изменения свойств и характеристик медицинского изделия, влияющих на его безопасность, качество и эффективность, или совершенствует его свойства и характеристики при неизменности функционального назначения и (или) принципа действия медицинского изделия;
- оригинал регистрационного удостоверения (дубликат);
- документы производителя и (или) организаций, осуществляющих проведение технических испытаний, токсикологических исследований, клинических испытаний (результаты соответствующих испытаний), подтверждающие, что внесение заявленных изменений не влечет изменения свойств и характеристик, влияющих на качество, эффективность и безопасность медицинского изделия, или совершенствует свойства и характеристики при неизменности функционального назначения и (или) принципа действия медицинского изделия, а также позволяющие оценить применяемые методы (методики) и перечень используемого испытательного оборудования;
- опись документов с указанием раздела Особенностей (раздел IV), в соответствии с которым планируется прохождение процедуры внесения изменений в документы, содержащиеся в регистрационном досье.
В случае, если указанные документы составлены на иностранном языке, они представляются с заверенным в установленном порядке переводом на русский язык.
Сроки принятия решения о внесении изменений в документы, содержащиеся в регистрационном досье для регистрационного удостоверения, оформленного в соответствии с Особенностями или Правилами, приведены в таблице 2.

Необходимо отметить, что в соответствии с п. 30 раздела IV Особенностей, возможно осуществление процедуры внесения изменений в документы, содержащиеся в регистрационном досье медицинского изделия отечественного производства, прошедшего государственную регистрацию не только в соответствии с Особенностями, но и в соответствии с Правилами. При внесении изменений в бланк регистрационного удостоверения, оформленного в соответствии с Правилами («бессрочного»), выдается также «бессрочное» регистрационное удостоверение.
Для отечественных производителей медицинских изделий Особенностями предусмотрена возможность при внесении изменений, связанных со сведениями о покупных изделиях, сырье, материалах и комплектующих и о составных частях, запасных частях и принадлежностях, сократить сроки проведения экспертизы до 5 рабочих дней с возможностью представления собственных результатов испытаний.
Кроме того, введение Особенностями положения, позволяющего производителю (изготовителю) или уполномоченному представителю производителя (изготовителя) заверять документы производителя (изготовителя) (при условии наличия и представления в Росздравнадзор документа, подтверждающего полномочия по заверению документов и вступившего в силу до оформления документов), также снижает сроки подготовки регистрационного досье в связи с отсутствием необходимости заверения документов в стране производителя.
Таким образом, Постановлением сформированы подходы к процедуре внесения изменений в регистрационное досье медицинского изделия, позволяющие сократить сроки принятия решения о внесении изменений в документы, содержащиеся в регистрационном досье, по сравнению с Правилами.