КОНТРОЛЬ КАЧЕСТВА И БЕЗОПАСНОСТИ
Развитие национальной системы мониторинга безопасности медицинских изделий. Усиление контроля за безопасностью медицинских изделий их производителями
Информация об авторах
1 — ФГБУ «Всероссийский научно-исследовательский и испытательный институт медицинской техники» Росздравнадзора, Российская Федерация, 115478, Москва, Каширское шоссе, д. 24, стр. 16.
2 — Министерство здравоохранения Российской Федерации, 107045, Российская Федерация, г. Москва, Колокольников переулок, д. 21.
3 — ФГБУ «Всероссийский научно-исследовательский и испытательный институт медицинской техники» Росздравнадзора, Российская Федерация, 115478, Москва, Каширское шоссе, д. 24, стр. 16.
4 — ФГБУ «Всероссийский научно-исследовательский и испытательный институт медицинской техники» Росздравнадзора, Российская Федерация, 115478, Москва, Каширское шоссе, д. 24, стр. 16.
5 — ФГБУ «Всероссийский научно-исследовательский и испытательный институт медицинской техники» Росздравнадзора, Российская Федерация, 115478, Москва, Каширское шоссе, д. 24, стр. 16.
Опубликовано: 25.06.2021
В статье приведен обзор ключевых изменений порядка мониторинга безопасности медицинских изделий в Российской Федерации и выполнено сопоставление национальных правил с Правилами проведения мониторинга безопасности медицинских изделий в рамках ЕАЭС. Рассмотрена роль производителя в системе мониторинга безопасности медицинских изделий.
Ключевые слова: медицинские изделия, безопасность медицинских изделий, неблагоприятное событие, правила мониторинга безопасности в рамках ЕАЭС, пострегистрационный клинический мониторинг
Введение
С 1 января 2021 г., в связи с вступлением в действие приказов Минздрава России от 15.09.2020 № 980н1 и от 19.10.2020 № 1113н2, утратили силу ранее действующие правила мониторинга безопасности медицинских изделий3.
Вступившие в силу новые национальные нормативные правовые акты гармонизированы с регламентирующими документами Евразийской экономической комиссии (далее – ЕЭК) в части мониторинга безопасности медицинских изделий (Решение Коллегии Евразийской экономической комиссии от 22.12.2015 № 174 «Об утверждении Правил проведения мониторинга безопасности, качества и эффективности медицинских изделий», далее – Решение Коллегии ЕЭК № 174), а их отличительной особенностью от ранее действующих правил на территории Российской Федерации является увеличение роли производителей медицинских изделий или их уполномоченных представителей при проведении мониторинга безопасности медицинских изделий (далее – мониторинг), а следовательно – их ответственности и степени вовлеченности в процесс.
В статье рассмотрены ключевые изменения в национальной системе мониторинга безопасности медицинских изделий и проведено их сопоставление с Правилами проведения мониторинга безопасности медицинских изделий в рамках Евразийского экономического союза (далее – ЕАЭС).
Определение термина «неблагоприятное событие» и другие понятия
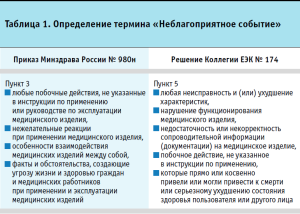
В новых правилах введено определение «неблагоприятное событие», перешедшее из целей мониторинга безопасности медицинских изделий в Российской Федерации, установленных приказом Минздрава России от 14.09.2012 № 175н. Однако оно отличается от определения неблагоприятного события в соответствии с Решением Коллегии ЕЭК № 174, в котором дополнительно включено определение некорректности или недостаточности сопроводительной документации на медицинское изделие (табл. 1). Следует также отметить, что в Правилах мониторинга в рамках ЕАЭС отдельно выделяется термин «нежелательное событие». Таким образом, правилами мониторинга в рамках ЕАЭС разграничиваются события при применении медицинских изделий, которые связаны с отклонениями функционирования медицинского изделия от ожидаемого либо с недостатками документации, и с непрогнозируемым исходом для пациента при «нормальной» работе изделия. Вместе с тем, независимо от принятого определения «неблагоприятного события», мониторинг безопасности на национальном уровне и в рамках ЕАЭС направлен на обеспечение безопасности пользователей медицинских изделий и предотвращения причинения вреда здоровью.
Роли участников процесса мониторинга безопасности
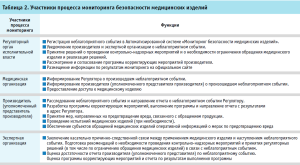
Действующими правилами мониторинга безопасности медицинских изделий в Российской Федерации и в рамках ЕАЭС установлены основные участники процесса мониторинга, определены их функции и задачи (табл. 2).
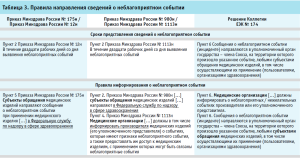
Отличительной особенностью нового приказа Минздрава России от 19.10.2020 № 1113н и Правил мониторинга безопасности в ЕАЭС является то, что медицинские организации в первую очередь должны информировать о произошедшем неблагоприятном событии производителя (или его уполномоченного представителя) и предоставлять ему доступ к медицинскому изделию, вовлеченному в инцидент, для проведения расследования, а производитель (уполномоченный представитель производителя) – направлять в регулирующий орган отчеты по результатам рассмотрения неблагоприятного события (табл. 3).
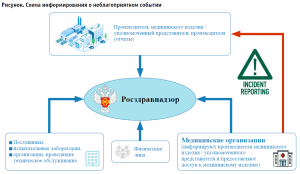
Следует отметить, что порядок проведения мониторинга безопасности медицинских изделий как в национальной системе, так и в рамках ЕАЭС, обязывает уведомлять регуляторный орган о выявленном неблагоприятном событии всех субъектов обращения, в том числе производителя или уполномоченного представителя производителя (см. рис.).
Часть отчета о неблагоприятном событии производителя идентична форме подачи сообщения о неблагоприятном событии, таким образом производитель может информировать регулирующий орган о неблагоприятном событии путем подачи отчета, что не противоречит требованиям действующих в Российской Федерации и на территории ЕАЭС нормативных правовых актов.
Источники информации о неблагоприятных событиях
В новых национальных правилах мониторинга безопасности расширены источники информации о проблемах безопасности медицинских изделий: если ранее в соответствии с национальным законодательством это был пассивный сбор сообщений от субъектов обращения медицинских изделий, а также активный – полученный в ходе проведения государственного контроля за обращением медицинских изделий, то теперь в качестве источника информации рассматриваются отчеты по безопасности производителя медицинских изделий или его уполномоченного представителя, включая периодические сводные отчеты и отчеты о пострегистрационным клиническом мониторинге.
В национальных правилах дополнительно рассматриваются сведения о проблемах безопасности медицинских изделий, полученные по результатам мониторинга публикаций на сайтах зарубежных регуляторных органов в сфере обращения медицинских изделий. Также в правилах мониторинга безопасности медицинских изделий сделан акцент на обеспечение усиленного наблюдения за имплантированными медицинскими изделиями, в том числе на отдаленном этапе имплантации. Для данной группы медицинских изделий национальным порядком мониторинга безопасности дополнительно предусматривается сбор и анализ данных, полученных из подсистем ведения специализированных регистров пациентов (табл. 4).

Требования к производителю при проведении мониторинга безопасности медицинских изделий
Новые порядки проведения мониторинга безопасности медицинских изделий в Российской Федерации, как и правила проведения мониторинга безопасности в рамках ЕАЭС, предусматривают обязанность производителя разрабатывать и проводить корректирующие мероприятия в целях устранения причины неблагоприятного события. Результатом выполнения корректирующих мероприятий является уведомление по безопасности, которое размещается на сайте регулирующего органа для уведомления субъектов обращения медицинских изделий о проводимых мероприятиях. Обязанности производителей и их уполномоченных представителей, виды отчетов и сроки их направления представлены в таблице 5.
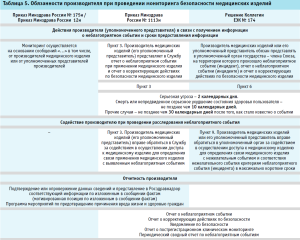
В соответствии с Решением Коллегии ЕЭК № 174, под корректирующим действием понимается действие, предпринятое производителем медицинских изделий с целью устранения причины обнаруженного несоответствия или нежелательного события). Это мероприятия производителя на месте производства, которые могут затрагивать технологические процессы, изменение конструкции медицинских изделий, используемых материалов, сопроводительной документации и т.д.
Как следует из определения, приведенного в Решении Коллегии ЕЭК № 174, корректирующее действие по безопасности медицинского изделия – это действие, предпринятое производителем медицинских изделий с целью снижения риска смерти или серьезного ухудшения состояния здоровья пользователей или третьих лиц, связанное с применением медицинского изделия. То есть корректирующие действия по безопасности направлены на предотвращение причинения вреда здоровью в связи с выявленной проблемой на местах применения медицинских изделий (у пользователей) и могут включать в себя добровольный отзыв медицинских изделий (партии, серии, модели), дополнительные рекомендации для обеспечения безопасности применения медицинских изделий, проведение ремонта на местах, настройки оборудования и прочее.
Уведомление по безопасности является сообщением, направленным производителем медицинского изделия или его уполномоченным представителем субъектам обращения медицинского изделия в связи с корректирующим действием по безопасности медицинского изделия, и информирует пользователей медицинских изделий о необходимых действиях для минимизации риска применения медицинского изделия.
Правила мониторинга безопасности медицинских изделий на национальном уровне и в рамках ЕАЭС предусматривают условия, когда производитель / уполномоченный представитель производителя может не представлять отчеты о неблагоприятных событиях. Например, отчеты могут не предоставляться по каждому неблагоприятному событию в случае, если проведены корректирующие действия и предоставлено уведомление для пользователей, а также по каждому неблагоприятному событию, в случае если неблагоприятное событие обозначено в анализе рисков, связанных с медицинским изделием и по ним представлены отчеты в регулирующий орган. Следует отметить, что в этих случаях требуется согласовать возможность представления сводных отчетов, а также их содержание и сроки (периодичность) направления.
Пострегистрационный клинический мониторинг
Нововведением законодательства в сфере мониторинга безопасности медицинских изделий в Российской Федерации и на территории ЕАЭС является требование к производителю медицинских изделий о проведении мониторинга безопасности и клинической эффективности медицинских изделий класса потенциального риска применения 3, а также имплантируемых в организм человека медицинских изделий классов потенциального риска применения 2б и 3 на пострегистрационном этапе в течение трех лет после регистрации медицинских изделий (табл. 6).

На основании определений, представленных в Правилах проведения клинических и клинико-лабораторных испытаний (исследований) медицинских изделий, утвержденных Решением Совета Евразийской экономической комиссии от 12.02.2016 № 29, мониторинг безопасности и клинической эффективности медицинских изделий после регистрации (или пострегистрационный клинический мониторинг) – это сбор, анализ и систематизация сведений на пострегистрационном этапе обращения медицинского изделия в целях доказательства его эффективности и безопасности на основании полученных клинических данных. В рамках клинической оценки/оценки эффективности проводится непосредственно сбор клинических данных, обзор ранее опубликованных данных, а также проводятся необходимые клинические исследования или исследования эффективности.
Клинический мониторинг проводится в соответствии с планом, который должен содержать цели и задачи, а также схему клинического мониторинга. Цели и задачи клинического мониторинга основываются на анализе имеющихся клинических данных, специфических особенностей и факторов риска, связанных с медицинским изделием. Аналогичное требование уже установлено в Европейском союзе (Regulation (EU) 2017/745 of the European Parliament and of the Council of 5 April 2017), а усиление наблюдения за медицинскими изделиями на пострегистрационном этапе обращения является общепринятой мировой практикой.
Заключение
Изменение правил проведения мониторинга безопасности медицинских изделий обуславливает большую вовлеченность производителя медицинского изделия и его уполномоченного представителя в процесс наблюдения за медицинскими изделиями после выпуска в гражданский оборот, что в свою очередь определяет их повышенную ответственность в случае непринятия своевременных мер по обеспечению безопасного применения медицинских изделий, непредставления сведений, предусмотренных правилами мониторинга безопасности медицинских изделий, или представления заведомо ложной информации. Для соответствия новым правилам мониторинга безопасности медицинских изделий от производителя может потребоваться адаптация системы менеджмента качества медицинских изделий, например, внесение изменений в процессы корректирующих и предупреждающих действий и процессы, связанные с потребителем. Вместе с тем, учитывая гармонизацию правил проведения мониторинга безопасности медицинских изделий на пострегистрационном этапе обращения в Российской Федерации и в рамках ЕАЭС снижаются затраты ресурсов производителя, необходимые для обеспечения соответствия правилам конкретного регулирующего органа.
_______________________________________________________________________
1 Приказ Минздрава России от 15.09.2020 № 980н «Об утверждении порядка мониторинга безопасности медицинских изделий».
2 Приказ Минздрава России от 19.10.2020 № 1113н «Об утверждении Порядка сообщения субъектами обращения медицинских изделий обо всех случаях выявления побочных действий, не указанных в инструкции по применению или руководстве по эксплуатации медицинского изделия, о нежелательных реакциях при его применении, об особенностях взаимодействия
медицинских изделий между собой, о фактах и об обстоятельствах, создающих угрозу жизни и здоровью граждан и медицинских работников при применении и эксплуатации медицинских изделий».
3 Утвержденные приказом Минздрава России от 14.09.2012 № 175н «Об утверждении Порядка осуществления мониторинга безопасности медицинских изделий» и приказом Минздрава России от 20.06.2012 № 12н «Об утверждении Порядка сообщения субъектами обращения медицинских изделий обо всех случаях выявления побочных действий, не указанных в инструкции по применению или руководстве по эксплуатации медицинского изделия, о нежелательных реакциях при его применении, об особенностях взаимодействия медицинских изделий между собой, о фактах и об обстоятельствах, создающих угрозу жизни и здоровью граждан и медицинских работников при применении и эксплуатации медицинских изделий».