ОХРАНА ЗДОРОВЬЯ МАТЕРИ И РЕБЕНКА, ВОПРОСЫ СЕМЬИ И РЕПРОДУКТИВНОГО ЗДОРОВЬЯ
Практический опыт диагностики редких наследственных болезней в рамках реализации программы расширенного неонатального скрининга
Информация об авторах
1 — Федеральное государственное автономное учреждение «Национальный медицинский исследовательский центр здоровья детей» Министерства здравоохранения Российской Федерации, 119296, Российская Федерация, г. Москва, Ломоносовский проспект, д. 2, стр. 1.
2 — Федеральное государственное автономное учреждение «Национальный медицинский исследовательский центр здоровья детей» Министерства здравоохранения Российской Федерации, 119296, Российская Федерация, г. Москва, Ломоносовский проспект, д. 2, стр. 1.
3 — Федеральное государственное автономное учреждение «Национальный медицинский исследовательский центр здоровья детей» Министерства здравоохранения Российской Федерации, 119296, Российская Федерация, г. Москва, Ломоносовский проспект, д. 2, стр. 1.
4 — Федеральное государственное автономное учреждение «Национальный медицинский исследовательский центр здоровья детей» Министерства здравоохранения Российской Федерации, 119296, Российская Федерация, г. Москва, Ломоносовский проспект, д. 2, стр. 1.
5 — Федеральное государственное автономное учреждение «Национальный медицинский исследовательский центр здоровья детей» Министерства здравоохранения Российской Федерации, 119296, Российская Федерация, г. Москва, Ломоносовский проспект, д. 2, стр. 1.
6 — Федеральное государственное автономное учреждение «Национальный медицинский исследовательский центр здоровья детей» Министерства здравоохранения Российской Федерации, 119296, Российская Федерация, г. Москва, Ломоносовский проспект, д. 2, стр. 1.
7 — Федеральное государственное автономное учреждение «Национальный медицинский исследовательский центр здоровья детей» Министерства здравоохранения Российской Федерации, 119296, Российская Федерация, г. Москва, Ломоносовский проспект, д. 2, стр. 1.
8 — Федеральное государственное автономное учреждение «Национальный медицинский исследовательский центр здоровья детей» Министерства здравоохранения Российской Федерации, 119296, Российская Федерация, г. Москва, Ломоносовский проспект, д. 2, стр. 1.
9 — Федеральное государственное автономное учреждение «Национальный медицинский исследовательский центр здоровья детей» Министерства здравоохранения Российской Федерации, 119296, Российская Федерация, г. Москва, Ломоносовский проспект, д. 2, стр. 1.
10 — Федеральное государственное автономное учреждение «Национальный медицинский исследовательский центр здоровья детей» Министерства здравоохранения Российской Федерации, 119296, Российская Федерация, г. Москва, Ломоносовский проспект, д. 2, стр. 1.
Опубликовано: 20.02.2025
Неонатальный скрининг является наиболее эффективным инструментом для выявления редких заболеваний в практике общественного здравоохранения во всем мире. В статье представлены результаты скрининга 324 734 новорожденных из 22 субъектов ЦФО и ДФО, осуществляемого в рамках выполнения федеральной программы «Расширенный неонатальный скрининг» с 01 января 2023 года в Медико-генетическом центре ФГАУ «НМИЦ здоровья детей» Минздрава России.
Охват скрининга составил 97,6% от числа всех новорожденных, сформированная группа риска составила 1,1% от числа всех обследованных, подтверждающая диагностика проведена для 96,3% детей, попавших в группу риска. Выявлено 215 больных детей, среди которых у 121 – наследственные болезни обмена, у 47 – спинальная мышечная атрофия, у 47 – первичные иммунодефициты. Наибольшая частота наследственных заболеваний была обнаружена в Тамбовской области – 1 случай на 749 новорожденных, а наименьшая – в Костромской области – 1 случай на 7398 новорожденных. Мы также выявили три случая дефицита 3-метилкротонил-КоА-карбоксилазы и один случай этилмалоновой энцефалопатии. Эти редкие заболевания не входят в список скринируемых и могут быть отнесены к случайным находкам расширенного неонатального скрининга.
Кроме того, нам удалось обнаружить патогенные биаллельные варианты у двух детей с глутаровой ацидурией, тип 2 и дефицитом митохондриального трифункционального белка, летальный исход которых наступил до проведения подтверждающей диагностики. Для оптимизации молекулярно-генетической диагностики в рамках расширенного неонатального скрининга было проведено сравнение аналитических характеристик наборов реактивов «TK-SMA» и НеоСкрин SMA/TREC/KREC и определение стабильности получаемых данных. Аналитические характеристики наборов реактивов были схожи, при этом оба набора имели хорошее воспроизведение результатов, независимо от оператора, который проводил исследование. Было показано, что средние значения KREC для наборов реактивов TK-SMA и Неоскрин статистически значимо не различались, в то время как средние значения TREC для двух наборов имели статистические значимые различия.
Ключевые слова: программа расширенного неонатального скрининга, новорожденные с наследственными заболеваниями, скрининг новорожденных, молекулярно-генетическая диагностика, биохимическая диагностика, наследственные болезни обмена (НБО), спинальная мышечная атрофия (СМА), первичные иммунодефициты (ПИД), тяжелая комбинированная иммунная недостаточность (ТКИН)
Введение
Орфанными болезнями в РФ принято считать заболевания, имеющие распространенность, не превышающую 10 случаев на 100 тысяч населения1, тогда как средняя мировая распространенность составляет 40 случаев на 100 тысяч человек [1]. Несмотря на относительно низкую распространенность, редкие болезни весьма многочисленны, их совокупный вклад в статистику инвалидности и смертности, в том числе младенческой, очень велик. В настоящее время известно более 10 тысяч редких болезней, 80% из них имеют идентифицированное генетическое происхождение и поражают от 3 до 4% новорожденных, 30% из которых не доживают до 5 лет [2–4]. Еженедельно в мировой научной литературе появляется описание от одного до пяти новых заболеваний, однако орфанные препараты или орфанные лекарственные средства существуют лишь для 3% этих болезней [5]. Большинство редких наследственных заболеваний манифестируют в детском возрасте, а значит актуальность их ранней диагностики сложно переоценить.
Неонатальный скрининг является наиболее эффективным инструментом для выявления редких наследственных заболеваний в практике общественного здравоохранения во всем мире. Это методологически сложный процесс, требующий серьезных усилий команды специалистов различного профиля, а также контроля и мониторинга со стороны федеральных органов исполнительной власти для достижения максимальной эффективности.
Внедрение программ неонатального скрининга, охватывающих орфанные болезни, для которых разработаны эффективные методы патогенетической терапии, позволяют значительно снизить инвалидизацию и младенческую смертность, тем самым значимо сокращая социально-экономические затраты на современное здравоохранение [6].
Термин «скрининг» с точки зрения методологии был предложен в 1968 году известными учеными в области медицины Уилсоном и Джангнером в монографии «Принципы и практика скрининга на выявление заболеваний», опубликованной по заказу ВОЗ [7]. В контексте монографии термин «скрининг» обозначал обнаружение недиагностированной ранее болезни с помощью набора определенных процедур или тестов. История неонатального скрининга насчитывает уже более 60 лет. Основоположником в этой области является микробиолог Роберт Гатри, который в 1962 году разработал лабораторный метод диагностики фенилкетонурии (ФКУ) [8], а в 1963 году в штате Массачусетс (США) стартовала первая программа скрининга новорожденных на ФКУ. Стоит отметить, что для проведения этого скрининга был использован биоматериал в виде пятен крови, высушенных на хроматографической бумаге.
Список тестируемых болезней постепенно расширялся, например, в 1970-е в программу скрининга в США была включена серповидно-клеточная анемия и врожденный гипотиреоз, в 1980-е годы – галактоземия, болезнь с запахом кленового сиропа мочи, врожденная гиперплазия надпочечников и биотинидазная недостаточность.
Новая эпоха в неонатальном скрининге, наступившая в 1990-е годы, была ознаменована созданием и внедрением в программы скрининга технологии тандемной масс-спектрометрии (МС/МС), которая позволила не только значимо ускорить время проведения исследований и повысить их эффективность, но и расширить список скринируемых заболеваний, для которых существовала патогенетическая терапия. Технология МС/МС и в наши дни является тестом первого уровня для диагностики наследственных болезней обмена в различных программах скрининга новорожденных во многих странах, однако число и спектр скринируемых заболеваний имеют существенные различия.
Наследственные болезни обмена (НБО) – гетерогенная группа редких наследственных заболеваний, насчитывающая на сегодняшний день более 1000 нозологических форм и занимающая одно из ведущих мест среди генетически детерминированных заболеваний человека [9]. Подавляющее большинство НБО наследуется по аутосомно-рецессивному типу и встречаются крайне редко, однако их суммарная частота в популяции составляет 1:1000–1:5000, затрагивая в целом до 3% населения земного шара [10].
Использование технологии МС/МС, а также успешное применение патогенетической терапии позволило значительно расширить спектр скринируемых НБО: аминоацидопатий, органических ацидурий, дефектов митохондриального бетаокисления жирных кислот, а в последние годы и лизосомных болезней накопления (ЛБН) вплоть до 50 различных нозологических форм.
Использование методов молекулярногенетической диагностики, в том числе ПЦР в режиме реального времени в качестве теста первого уровня в программах неонатального скрининга показало высокую эффективность для заболеваний, не имеющих специфичных диагностических биохимических маркеров. Одним из ярких примеров является спинальная мышечная атрофия (СМА), неонатальный скрининг которой, начиная с 2018 года, успешно осуществляется в ряде штатов США. Еще одним прорывом в области диагностики и ранней терапии редких болезней стало включение в программы неонатального скрининга первичных иммунодефицитов.
Врожденные дефекты иммунитета или первичные иммунодефициты (ПИД) – группа генетически детерминированных заболеваний, приводящих к дефектам различных звеньев иммунитета[11]. Одним из заболеваний данной группы является тяжелая комбинированная иммунная недостаточность (ТКИН), связанная с отсутствием, либо сниженным числом Т-лимфоцитов, зачастую приводящая к гибели пациентов на первом году жизни, обусловленной тяжелыми рецидивирующими инфекциями. Для своевременной постановки диагноза и успешной терапии данной группы заболеваний, в том числе на доклинической стадии, применяются различные программы скрининга новорожденных на ТКИН [12]. Большинство из них основано на анализе содержания молекулярного маркера – Т-рецепторных эксцизионных колец (T-cell receptor excision circles, TREC) в сухих пятнах крови.
Возможность использования TREC как маркера ПИД обсуждалась еще в конце 1990-х годов, однако активное внедрение данного метода в клиническую практику началось лишь в начале 2000-х, когда для диагностики стали использовать сухие пятна крови, а уже в начале 2010-х годов определение TREC стало стандартным методом исследования в программах неонатального скрининга в США [13], а позднее и в других странах [14].
В свою очередь, было показано, что Каппа-рекомбинационные эксцизионные кольца (KREC) могут выступать в качестве маркера В-клеточного созревания [15]. Использование этого маркера в программах скрининга позволяет успешно диагностировать пациентов с такими нарушениями созревания В-лимфоцитов, как агаммаглобулинемия [16]. В настоящее время KREC используется в программах неонатального скрининга в немногих странах, таких как Польша, Швеция и Испания, при этом в некоторых из них, в том числе в программе расширенного неонатального скрининга в России, осуществляющейся с 01 января 2023 года, практикуется мультиплексный анализ TREC/KREC [17].
Включение тестов на ПИД в программы неонатального скрининга позволяет значимо ускорить процесс постановки диагноза для данной группы пациентов еще на доклинической стадии, тем самым существенно снижая высокую летальность пациентов в случае отсутствия корректного диагноза и своевременного назначения терапии [18].
Стоит отметить, что, несмотря на то, что к настоящему моменту в США 38 болезней включены в единый рекомендованный список заболеваний для скрининга (англ. Recommended uniform screening panel, RUSP), в этой стране не существует единой федеральной программы, а программы неонатального скрининга различаются в зависимости от штата. Так, например, только в январе 2024 года спинальная мышечная атрофия была включена в панель нозологий, подлежащих неонатальному скринингу на Гавайях [19].
Федеральная программа неонатального скрининга отсутствует и в Канаде, где число нозологий варьирует на разных территориях. В странах Тихоокеанского региона, в частности, в Австралии и Новой Зеландии также существуют программы неонатального скрининга. В Австралии скрининг проводится с 2005 года и охватывает 25 заболеваний [20].
В Российской Федерации программы неонатального скрининга насчитывают около 40 лет. С 1986 года скринируется ФКУ, в 1993 году был добавлен врожденный гипотериоз, с 2006 года число скринируемых нозологий пополнилось еще тремя: галактоземией, муковисцидозом и адреногенитальным синдромом.
Согласно Распоряжению Правительства Российской Федерации от 09.06.20222 №1510-р, а также приказу Минздрава России от 27.12.2022 № 808н3, ФГАУ «НМИЦ здоровья детей» Минздрава России было доверено осуществление программы расширенного неонатального скрининга новорожденным из 22 субъектов Центрального и Дальневосточного Федеральных округов Российской Федерации.
Цель работы
Целью данного исследования является описание результатов расширенного неонатального скрининга детей, родившихся в 2023 и 2024 годах в 22 субъектах РФ, биоматериал которых был доставлен в Медикогенетический центр ФГАУ «НМИЦ здоровья детей» Минздрава России, а также описание сравнения аналитических характеристик наборов реактивов для молекулярно-генетической диагностики СМА и ПИД.
Материалы и методы
С 01 января 2023 года по 31 декабря 2024 года в Медико-генетический центр ФГАУ «НМИЦ здоровья детей» Минздрава России (Центр 3А) поступило 324 734 образца новорожденных в виде пятен крови, высушенных на фильтровальной бумаге, из 22 субъектов РФ: Амурской Белгородской, Брянской, Владимирской, Воронежской, Ивановской, Калужской, Костромской, Курской, Липецкой, Орловской, Рязанской, Сахалинской, Смоленской, Тамбовской, Тверской, Тульской, Ярославской областей, Камчатского, Приморского и Хабаровского края, а также Еврейской автономной области. Молекулярно-генетическая диагностика ПИД и СМА проводилась методом мультиплексной ПЦР в режиме реального времени с использованием реактивов4, биохимическая диагностика наследственных болезней обмена проводилась методом тандемной масс-спектрометрии (ТМС) с использованием набора реактивов для недериватизированной ТМС. Подтверждающая диагностика в рамках РНС выполнялась в ФГБНУ «Медикогенетический научный центр им. акад. Н.П. Бочкова» (Центр 3Б). Подтверждающая диагностика в рамках научно-исследовательской работы проводилась в лаборатории медицинской геномики Медико-генетического центра ФГАУ «НМИЦ здоровья детей» Минздрава России. Информированные добровольные согласия на проведение исследования были подписаны законными представителями (родителями) всех обследованных детей.
Молекулярно-генетическая диагностика проходила в два этапа: выделение ДНК и последующая мультиплексная ПЦР в режиме реального времени. Выделение ДНК проводилось на системе для автоматического выделения и очистки нуклеиновых кислот из биологического материала, согласно рекомендациям фирмыпроизводителя реактивов. Последующая ПЦР в режиме реального времени проводилась на детектирующих амплификаторах. Первичная обработка данных проводилась в программном обеспечении с последующей интерпретацией согласно алгоритму, разработанному в лаборатории.
Количественное определение и анализ концентраций аминокислот, сукцинилацетона и ацилкарнитинов проводились с использованием реагентов для недериватизированной тандемной масс-спектрометрии на тандемных масс-спектрометрах в образцах сухих пятен крови. Подготовка образцов, этапы экстракции и инжекции проводились согласно рекомендациям фирмы-производителя реактивов. Обработка данных проводилась входящим в состав системы скрининга программным обеспечением. Пограничные значения доверительного интервала для аналитов были определены путем расчета концентрации аналитов, соответствующих 0.5-му и 99.5-му перцентилям.
Газовую хроматографию образцов мочи проводили на газовом хроматографе с масс-селективным детектором.
В качестве подтверждающего метода валидации гомозиготной делеции экзона 7 гена SMN1, а также определения числа копий гена SMN2 у пациентов со СМА использовалась мультиплексная лигазозависимая цепная реакция (MLPA) с применением набора реактивов. Реакцию проводили согласно рекомендациям фирмы-производителя, а последующий фрагментный анализ осуществлялся на генетическом анализаторе.
Для поиска генетических причин иных заболеваний проводилось высокопроизводительное секвенирование. Геномные библиотеки были приготовлены с использованием набора реактивов, согласно рекомендациям фирмы-производителя. Обогащение библиотек целевыми областями исследуемых генов проводили при помощи гибридизационных проб. Секвенирование осуществлялось на генетических анализаторах. Анализ данных осуществлялся в соответствии c рекомендациями GATK Best Practices (https://gatk.broadinstitute.org/). Анализ патогенности выявленных миссенс-вариантов, не описанных ранее, осуществляли при помощи in silico анализа. Патогенность генетических вариантов, описанных ранее, проверяли в базе данных мутаций генома человека HGMD Professional. Нуклеотидные варианты, выявленные при помощи высокопроизводительного секвенирования, были валидированы методом секвенирования по Сэнгеру.
Результаты
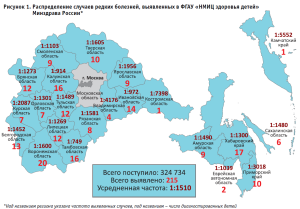
С 01 января 2023 года стартовала федеральная программа «Расширенный неонатальный скрининг», в рамках которой обследуются все новорожденные граждане РФ на 29 заболеваний нарушения обмена веществ, спинальную мышечную атрофию и первичные иммунодефициты. В Медикогенетическом центре ФГАУ «НМИЦ здоровья детей» Минздрава России за 2 года было обследовано 324 734 новорожденных, что составило 97,6% от числа детей, родившихся живыми в 22 субъектах РФ по данным Росстата (табл. 1), сформированная группа риска составила 1,097% от числа всех обследованных, подтверждающая диагностика проведена для 96,29% детей, попавших в группу риска. В результате выявлено 215 больных детей, среди которых у 121 ребенка обнаружены наследственные болезни обмена, у 47 – спинальная мышечная атрофия, у 47 – первичные иммунодефициты.
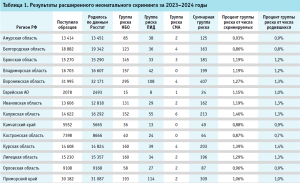
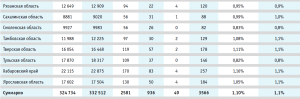
Общая частота новорожденных с наследственными заболеваниями из субъектов ЦФО и ДФО составила 1:1510, причем частота наследственных болезней обмена – 1:2683, а частота как ПИД, так и СМА – 1:6909 (рис. 1). Наибольшая частота была обнаружена в Тамбовской области – 1 случай на 749 новорожденных, а наименьшая – в Костромской области – 1 случай на 7 398 новорожденных.
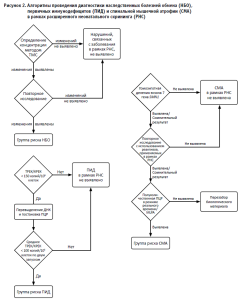
Опыт диагностики редких заболеваний в лабораториях Медико-генетического центра ФГАУ «НМИЦ здоровья детей» Мин здрава России [47], а также первые результаты РНС позволили сформировать и в дальнейшем скорректировать алгоритм диагностики НБО, СМА и ПИД (рис. 2).
В результате проведения расширенного неонатального скрининга на СМА было выявлено 47 новорожденных.
Наибольшее число случаев зафиксировано в Калужской области (6 случаев, частота составила 1:2437 новорожденных), в то время как наибольшая частота была зарегистрирована в Еврейской АО – 1 случай на 2078 новорожденных (ввиду наименьшего числа новорожденных). Ни одного ребенка со СМА не было выявлено во Владимирской, Костромской, Смоленской, Тульской областях и Камчатском крае. Медиана возраста детей со СМА на момент диагностики составила 6 дней.
Известно, что для дальнейшего лечения больных со СМА необходимо учитывать различные генетические модификаторы заболевания, основным из которых является число копий гена SMN2 [21].
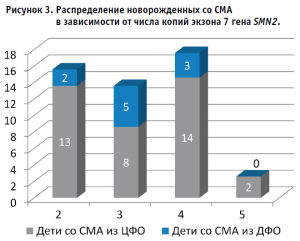
На рисунке 3 представлено распределение числа копий экзона 7 гена SMN2, выявленного нами у пациентов из ЦФО и ДФО. Процент пациентов, у которых было выявлено 4 и более копий гена SMN2 составляет 40,4%. Кроме того, было выявлено 5 (10,4% от всех подтвержденных случаев) новорожденных с химерным геном SMN2. Наиболее часто встречаемый вариант такого гена – SMN2 экзон 7 – 3 копии, SMN2 экзон 8 – 2 копии.
Из 47 больных СМА 39 детей уже получают патогенетическую терапию (45% онасемноген абепарвовек, 28% нусинерсен и 11% рисдиплам), 2 пациента находятся на динамическом наблюдении и 6 отказались от лечения.
В структуре первичных иммунодефицитов из 936 случаев, внесенных в группу риска, 880 (94,2%) детей были отправлены на подтверждающую диагностику. Анализ оставшихся 56 образцов указал на летальный исход в 45 случаях, отказ от проведения исследования – в 7 случаях, в 4 случаях родители с ребенком выехали в другой регион или за рубеж до взятия биоматериала для подтверждающей диагностики. Повторное снижение TREC и/ или KREC зафиксировано в 136 образцах (14,6%). Иммунофенотипирование выявило 47 пациентов с признаками иммунодефицитного состояния, у 20 из которых было обнаружено снижение TREC, у 23 – KREC, у 4 – снижение обоих параметров. Из 47 новорожденных с положительными результатами иммунофенотипирования 44 ребенка находятся под динамическим наблюдением, 6 из них получают патогенетическую терапию, в трех случаях зафиксирован летальный исход вскоре после проведения подтверждающей диагностики. Для оптимизации работы лаборатории было проведено определение стабильности получаемых данных разными наборами реактивов. Ретроспективно были определены медианные значения концентрации TREC и КREC, полученные разными операторами с использованием разных лотов реактивов. Данные представлены на рисунке 4.
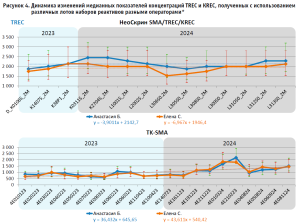
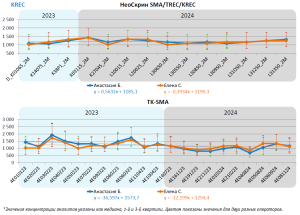
Было продемонстрировано адекватное воспроизведение результатов, независимо от оператора, проводившего исследование. Также было показано, что наборы Неоскрин имеют лучшую воспроизводимость независимо от лота, в то время как для наборов реактивов ТК СМА уровень TREC и KREC может значимо меняться в зависимости от лота. Кроме того, наблюдается тенденция к увеличению медианного показателя TREC для наборов реактивов ТК СМА. В декабре 2024 года медианное значение этого показателя составило 1488 копий/105 ядросодержащих клеток, в то время как в январе 2023 года оно было 762 копии/105 ядросодержащих клеток.
При этом аналитические характеристики наборов реактивов схожи и могут быть использованы для проведения РНС. Также было проведено сравнение доли образцов, попавших в группу риска ПИД для каждого из наборов реактивов (рис. 5).
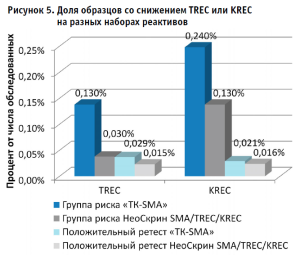
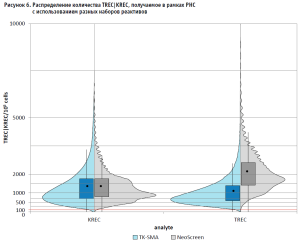
Было показано, что группа риска по TREC значимо различается между наборами. При использовании ТК СМА группа риска больше, как и доля подтвердившихся при повторном исследовании от общего числа обследованных. Это можно объяснить тем, что среднее значение TREC, определяемое наборами реактивов Неоскрин, в 1,8 раза больше, нежели наборами реактивов ТК СМА (рис. 6).
Совокупность данных, полученных при изучении аналитических характеристик наборов реактивов, повлияла на принятие решения о необходимости проведения повторного исследования с того же пятна крови для образцов со значениями TREC|KREC меньше 150 копий на 105 ядросодержащих клеток. Это позволило не пропустить двух пациентов, у которых концентрация TREC и KREC при первичном тесте была выше 100 копий на 105 ядросодержащих клеток, а в последующем их диагноз был подтвержден на ИФТ. В структуре НБО из 2579 случаев, внесенных в группу риска, материал 2502 (97,01%) детей был отправлен на подтверждающую диагностику. Анализ оставшихся 77 образцов указал на летальный исход в 21 случае, отказ от подтверждающего исследования – в 38 случаях, переезд в другой регион до взятия биоматериала на подтверждающую диагностику – в 18 случаях.
Молекулярно-генетическое исследование проведено 208 новорожденным с положительным результатом повторно проведенной диагностики аминокислот и ацилкарнитинов, биаллельные варианты выявлены у 121 новорожденного.
У 84 (69,4%) детей диагностирована гиперфенилаланинемия (ГФА), при этом подавляющее число случаев было обусловлено мутациями гена PAH (81/96,4%), тогда как лишь у 3 (3,6%) детей были выявлены мутации гена PTS. Во всех регионах, кроме Рязанской области, Камчатского края и Еврейской автономной области, был выявлен хотя бы 1 случай ГФА. В Ивановской области выявлено наибольшее число новорожденных – 10 с подтвержденным диагнозом ГФА, что составляет 12% от числа всех выявленных случаев этого заболевания. У 3 новорожденных из Ивановской, Смоленской и Воронежской областей выявлены биаллельные варианты в гене PTS (OMIM 612719), описанные у пациентов с ГФА с дефицитом тетрагидробиоптерина, тип А (OMIM 261640). Лидером по частоте заболеваний из группы НБО является Воронежская область. На ее долю приходится 9,6% от всех выявленных случаев ГФА. Наследственная тирозинемия, тип 1, выявлена у двух новорожденных, а болезнь «кленового сиропа» выявлена в трех случаях, причем два из них обнаружены в Воронежской области. В группе редких заболеваний, связанных с нарушением цикла образования мочевины, у двух новорожденных выявлена цитруллинемия, тип 1, и один случай аргининянтарной ацидурии. Шестеро новорожденных находятся на динамическом наблюдении, трое получают специализированную смесь, в двух случаях, несмотря на начатое лечение, избежать летального исхода не удалось.
Из заболеваний группы органических ацидурий были обнаружены 3 случая метилмалоновой ацидурии, глутаровая ацидурия, тип 1 – в двух случаях; по одному случаю – изовалериановой, пропионовой и глутаровой ацидурии, тип 2. Все младенцы из этой группы находятся на динамическом наблюдении и 6 дополнительно получают патогенетическую терапию в соответствии с утвержденными клиническими рекомендациями.
В группе заболеваний с нарушением окисления жирных кислот и обмена карнитина, недостаточность среднецепочечной ацил-КоА дегидрогеназы жирных кислот была выявлена у 15 новорожденных (1:21 649). У троих новорожденных выявлен дефицит ацил-КоА дегидрогеназы жирных кислот с очень длинной углеродной цепью, в одном случае – недостаточность длинноцепочечной 3-гидрокси-ацил-КоА дегидрогеназы жирных кислот. Трое новорожденных с подтвержденными диагнозами из группы нарушения бета-окисления жирных кислот поставлены под динамическое наблюдение и получают терапию, остальные находятся только на динамическом наблюдении – специализированное лечебное питание им в настоящее время не требуется, достаточно соблюдения интервалов кормления.
В ходе массового обследования новорожденных детей выявляются НБО, не включенные в программу РНС. Они встречаются крайне редко, фенотип этих детей варьирует от минимальных клинических проявлений вплоть до летального исхода. К подобным случайным находкам можно отнести нескольких выявленных нами детей. У одного из таких новорожденных был обнаружен высокий уровень показателя бутирилкарнитина, С4 (1,95 мкмоль/л при норме 0,09–0,89 мкмоль/л). В результате молекулярно-генетического исследования были выявлены патогенные биаллельные варианты в гене ETHE1 (chr19:43526608ACT>A и chr19:43526343G>A), мутации которого описаны у пациентов с ультраредкой этилмалоновой энцефалопатией (OMIM 602473).
У двух других новорожденных из ДФО были выявлены высокие уровни показателя 3-гидроксиизовалерилкарнитина (С5ОН) (10,2 мкмоль/л и 1,58 мкмоль/л, при норме 0,11–0,57 мкмоль/л). При повторном исследовании в рамках подтверждающей диагностики в центре 3Б также были выявлены высокие уровни данного показателя, после чего там же был проведен безуспешный поиск каузальных нуклеотидных вариантов в генах, мутации в которых вызывают развитие НБО, включенных в перечень скринируемых в рамках РНС. Принимая во внимание значения показателя С5ОН, значительно превышающие референсный интервал, нами было принято решение о поиске молекулярно-генетической причины этих завышений, в результате чего были выявлены биаллельные гетерозиготные варианты в первом случае chr5:71596278A>G и chr5:71656743G>T в гене MCCC2 (OMIM 609014); и chr5:71641018G>A в гомозиготном состоянии в гене MCCC2 во втором случае. Согласно базе данных OMIM, мутации в этом гене описаны у пациентов с дефицитом 3-метилкротонил-КоА-карбоксилазы, тип 2 (OMIM 210210), наследуемым по аутосомно-рецессивному типу.
Четвертый ребенок в возрасте 1 год и два месяца поступил в отделение патологии новорожденных детей ФГАУ «НМИЦ здоровья детей» с отрицательными результатами РНС. Исследование спектра аминокислот и ацилкарнитинов, проведенное нами, выявило значительное завышение показателя 3-гидроксиизовалерилкарнитина (С5ОН) до 54,03 мкмоль/л. В результате последующей молекулярно-генетической диагностики методом высокопроизводительного секвенрирования был выявлен патогенный нуклеотидный вариант chr3:183041679T>G в гомозиготном состоянии в гене MCCC1 (OMIM 609010), мутации в котором описаны у пациентов с дефицитом 3-метилкротонил-КоА-карбоксилазы, тип 1 (OMIM 210200), наследуемым по аутосомно-рецессивному типу.
Кроме того, нам удалось обнаружить причину ранних летальных исходов у двух пациентов с НБО, входящих в список выявляемых в рамках РНС. У новорожденного из Ярославской области по результатам ТМС были выявлены высокие уровни показателей 3-гидроксипальмитоилкарнитина (С16ОН) до 3,56 мкмоль/л (норма 0,01–0,09 мкмоль/л) и 3-гидроксистеароилкарнитина (С18ОН) до 1,14 мкмоль/л (норма 0,01–0,04 мкмоль/л). К сожалению, ребенок скончался до отправки биоматериала на подтверждающую диагностику в центр 3Б. По результатам молекулярно-генетического исследования нами был выявлен нуклеотидный вариант chr2:26285469AT>A в гомозиготном состоянии в гене HADHB (OMIM 143450), патологичные изменения в котором приводят к дефициту митохондриального трифункционального белка.
В результате исследования спектра аминокислот и ацилкарнитинов у второго новорожденного из Тамбовской области выявлен целый ряд завышенных показателей, характерных для глутаровой ацидурии, тип 2 (бутирилкарнитина С4 до 3,73 мкмоль/л; изовалерилкарнитина С5 до 3,86 мкмоль/л; гексаноилкарнитина С6 до 2,69 мкмоль/л; октаноилкарнитина С8 до 4,1 мкмоль/л; деканоилкарнитина С10 до 2,18 мкмоль/л; додеканоилкарнитина С12 до 2,89 мкмоль/л; тетрадеканоилкарнитина С14 до 5,11 мкмоль/л). Однако, ребенок скончался до отправки биоматериала на подтверждающую диагностику в центр 3Б. По результатам молекулярно-генетического исследования нами был выявлен патогенный нуклеотидный вариант chr4:158690393G>A в гомозиготном состоянии в гене ETFDH (OMIM 231675), мутации в котором приводят к множественному дефициту ацил-КоА-дегидрогеназы (глутаровой ацидурии, тип 2С) (OMIM 231680), наследуемому по аутосомно-рецессивному типу.
Кроме того, во ФГАУ «НМИЦ здоровья детей» Минздрава России госпитализируются дети без установленного диагноза, но с подозрением на болезни, не выявленные в рамках РНС. В октябре 2023 года на госпитализацию поступил ребенок из СКФО в возрасте 9 месяцев с регрессом ранее приобретенных навыков, задержкой психомоторного развития, вялостью. На МРТ головного мозга – картина атрофических изменений вещества больших полушарий и мозжечка, симметричного поражения белого вещества больших полушарий, подкорковых структур, гиппокампов, ствола мозга и мозжечка на фоне задержки миелинизации, расширения сильвиевых щелей. По результатам тандемной масс-спектрометрии нами было выявлено увеличение концентрации показателя глутарилкарнитина (C5DC) до 8,28 мкмоль/л (норма 0,04–0,69 мкмоль/л). По результатам газовой хроматографии органических кислот в моче выявлено увеличение концентрации показателей глутаровой кислоты до 3400 мМ/М креатинина (норма до 5 мМ/М креатинина) и 3-гидроксиглутаровой кислоты до 145 мМ/М креатинина (норма до 12 мМ/М креатинина). Учитывая данные анамнеза жизни и заболевания, клиническую картину, данные МРТ и лабораторных исследований у ребенка был заподозрен диагноз глутаровая ацидурия, тип 1. В результате молекулярно-генетического исследования нами был выявлен патогенный нуклеотидный вариант chr19:12896955G>A в гомозиготном состоянии в гене GCDH (OMIM 231670), что подтвердило диагноз глутаровой ацидурии, тип 1, не установленный в ходе расширенного неонатального скрининга.
Еще одним примером поздней диагностики орфанной болезни, входящей в перечень скринируемых в ходе реализации РНС, является пятимесячный ребенок из субъекта РФ, также не входящего в ЦФО и ДФО, родители которого обратились к генетику по месту жительства в связи с регрессом моторных навыков, что позволило заподозрить СМА и отправить биоматериал в нашу лабораторию в рамках программы селективного скрининга. В ходе проведенной молекулярно-генетической диагностики нами была выявлена гомозиготная делеция гена SMN1 и 2 копии гена SMN2, что подтвердило СМА.
Обсуждение
Согласно опубликованным в открытой медицинской печати данным, усредненная частота орфанных наследственных заболеваний, выявленных в России в ходе реализации программы РНС за 2023 год, составила 1:1818 новорожденных [188]. По результатам проведения РНС в 22 субъектах ЦФО и ДФО, частота редких наследственных болезней (НБО, ПИД и СМА) за 2023 и 2024 годы оказалась несколько выше и составила 1:1510. Это может быть связано как с более высокой частотой скринируемых нозологий в ЦФО и ДФО, что не было описано ранее, так и с более эффективным выявлением орфанной патологии.
Наибольшая суммарная частота НБО, перечень которых практически совпадает с перечнем нозологий, скринируемых в рамках РНС, описан в Корее (1:2000) и Германии (1:2517). Средняя же суммарная частота по миру составляет 1:20005000 [22]. Частота НБО в 22 регионах ЦФО и ДФО составила 1:2683. По нашим данным, наиболее часто выявляемыми заболеваниями за 2023 и 2024 годы стали ГФА (1:3865) и дефицит среднецепочечной ацил-КоА-дегидрогеназы жирных кислот (1:21649), что лишь отчасти коррелирует с результатами неонатального скрининга, полученными в Германии: ГФА (1:5262), дефицит среднецепочечной ацил-КоА-дегидрогеназы жирных кислот (1:10086) [23]. В спектре мутаций гена ACADM (607008) превалирует мажорный вариант c.985A>G, обнаруженный у 73% детей, причем у 40% – в гомозиготном состоянии, что также соответствует результатам зарубежных исследований [24]. Частота ГФА в 22 субъектах ЦФО и ДФО варьирует от 1:1361 в Ивановской области до 1:14608 в Курской области.
В спектре мутаций превалирует частый вариант c.1222C>T, выявленный у 51,25% новорожденных с вариантами в гене PAH (OMIM 612349). Этот вариант также является мажорным у европейцев (75–85%), особенно жителей стран Балтийского региона [25, 26]. При подтверждении диагноза ГФА важно незамедлительно начать лечение, чтобы достичь поддержания концентрации фенилаланина в рамках терапевтического диапазона (120–360 мкмоль/л) путем ограничения фенилаланина и натурального белка. Для этого разработаны специализированные смеси лечебного питания на основе аминокислот без фенилаланина с повышенным содержанием тирозина, которые компенсируют дефицит белка и других жизненно важных нутриентов. Частота других заболеваний из группы аминоацидопатий в 22 субъектах ЦФО и ДФО составляет 1:162 187 (наследственная тирозинемия, тип 1 и цитруллинемия, тип 1). По данным зарубежных источников, частота наследственной тирозинемии, тип 1, выявленной в результате неонатального скрининга, колеблется от 1:13 636 до 1:750 000 живых новорожденных [27], а цитруллинемии, тип 1 – 1:152 500 [28], что не противоречит нашим данным. Частота болезни «кленового сиропа» в 22 субъектах ЦФО и ДФО составляет 1:108 124, тогда как в мире эта цифра составила 1:185 000 новорожденных [29].
Частота глутаровой ацидурии, тип 1 варьирует от 1:15 060 в Австрии [30], 1:57 802 в Испании [31] до 1:135 000 в Германии [32]. Среди новорожденных, обследованных нами, было выявлено 2 случая глутаровой ацидурии, тип 1 (частота 1:162 187), что коррелирует с данными Германии. СМА – жизнеугрожающее заболевание, для которого крайне важна быстрая молекулярно-генетическая верификация диагноза и своевременное начало лечения [6]. РНС позволяет быстро выявить и начать лечение не только больных с тяжелой формой заболевания, первые симптомы которого появляются в неонатальном периоде, но и пациентов с более легкой формой, манифестация которой наступает позднее. Программы неонатального скрининга вносят существенные коррективы в распространенность любых скринируемых редких болезней, СМА не является исключением. Так, в США с 2016 по 2018 годы с использованием различных подходов было проанализировано 2 395 718 образцов новорожденных и выявлено 180 положительных, а также 10 ложноположительных случаев СМА. Тем самым частота с теоретических 1:13 310 сократилась до 1:11 236 [33, 34]. В России за 2023 год было проанализировано 1 227 130 образцов новорожденных и найдено 117 больных СМА – 1:10 480 новорожденных [18]. В то же время, по данным РНС, проведенного в 22 субъектах ЦФО и ДФО, частота СМА значительно превышает всероссийские данные и составляет 1:6909. Эти различия могут указывать как на ложноотрицательные случаи СМА, так и на недостаточный охват новорожденных расширенным неонатальном скринингом в отдельных регионах РФ. Так, при уровне охвата в 98,03% в 2023 году [18] не обследовано в рамках РНС более 20 000 новорожденных, среди которых могут быть 15 детей с различными орфанными заболеваниями, включая СМА. Кроме того, в 2023 году не было отправлено на подтверждающую диагностику 12,9% детей, внесенных в группу риска, что также снижает итоговую частоту скринируемых нозологий.
Результаты программы селективного скрининга на СМА, проводимой в нашей лаборатории, согласуются с зарубежными, демонстрируя превалирование 3 копий гена SMN2, однако, в рамках РНС в 22 субъектах ЦФО и ДФО мы обнаружили превалирование генотипа с 4 копиями экзона 7 гена SMN2 [35]. Такие результаты можно связать как с невключением детей с большим числом копий в программы раннего селективного скрининга ввиду отсутствия у них клинических проявлений заболевания, так и с возможными геногеографическими особенностями новорожденных из ЦФО и ДФО. В пользу последнего говорит тот факт, что по результатам первого года РНС в России наибольшее число пациентов имело 2 копии гена SMN2 [18]. Сопоставимая с нашими данными частота генотипов пациентов с 4 копиями гена SMN2 также описана в Германии. В результате неонатального скрининга в 2018 и 2019 годах было выявлено 38 детей, больных СМА, из которых 40% имели 4 и более копий гена SMN2 [36].
Частота другого модификатора заболевания – числа химерных вариантов гена SMN согласуется с общемировыми данными: 10,5% среди генотипированных новорожденных из 22 субъектов ЦФО и ДФО статистически не отличается от 10,8% в среднем по России [18] и от 10% в Нидерландах [37]. Наличие химерного варианта гена SMN обычно сопровождается более мягким фенотипом и лучшим прогнозом по лечению СМА [38, 39].
Выявление СМА в течение первых 6 суток жизни, которое удалось реализовать в ФГАУ «НМИЦ здоровья детей», должно значительно улучшить прогноз по лечению тяжелых форм болезни в случае его инициации в максимально сжатые сроки. По данным литературы, раннее начало терапии является ключевым предиктором благоприятного прогноза лечения [40].
В наше время известно более 400 различных нозологических единиц ПИД, обусловленных мутациями более чем в 330 генах [41]. Различные мутации в одном и том же гене могут приводить к различным фенотипам заболевания. Для скрининга этих болезней зачастую используют методы, позволяющие определить содержание TREС и KREC в крови пациентов. В России в рамках РНС принято оценивать этот показатель как количество копий TREC|KREC/105 ядросодержащих клеток. Такой подход распространен далеко не во всех странах. Зачастую используют другие метрики, например, количество копий в одном пятне или мл крови, в связи с этим могут возникать трудности в сравнении данных, полученных в рамках РНС с общемировыми. Другим важным нюансом является отрезная точка, используемая для диагностики ПИД. Хотя для каждого из наборов реактивов, используемых для скрининга, прописаны свои референсные значения, согласно принятым нормам в группу риска попадают только образцы, у которых концентрация TREC|KREC ниже 100 копий. Неонатальный скрининг способствовал выявлению частот ТКИН и врожденной агаммаглобулинемии в разных странах. В Израиле частота ПИД по результатам многолетнего неонатального скрининга составила 1:6463 [42], в остальных странах значительно реже: в Норвегии частота ТКИН составляет – 1:22 000 [43], в Испании – 1:130 000 [44], в США 1:58 000 новорожденных [45]. В 22 субъектах ЦФО и ДФО общая частота ПИД составила 1:6910 новорожденных, что согласуется с данными, полученными в Израиле.
В результате проводимой работы был модифицирован алгоритм диагностики НБО, ПИД и СМА в рамках РНС. Проведение повторного исследования с того же биологического материала при изменениях концентрации аналитов позволяет уменьшить долю ложноположительных результатов в случае НБО. Это способствует снижению эмоционального давления на семьи детей и сокращает материальные и трудовые ресурсы на этапе подтверждающей диагностики. В то же время, для диагностики ПИД и СМА повторное исследование для сомнительных результатов (СМА) и попадающих в так называемую серую зону (ПИД) позволяет уменьшить долю как ложноположительных, так и ложноотрицательных результатов. В рамках диагностики СМА были образцы, у которых при первичном исследовании диагностировали гомозиготную делецию экзона 7 гена SMN1. Однако, повторное исследование неизменно позволяло трактовать этот результат как ложноположительный, возможно возникающий при нарушениях преаналитического этапа, к примеру, кровь на фильтровальной бумаге могла содержать следы гепарина из катетера.
Результатами серьезной аналитической работы сотрудников нашего центра также явился проведенный анализ стабильности и воспроизводимости результатов определения ТRЕC и KREC с использованием различных наборов реактивов. Результаты таких исследований могут быть полезны с практической точки зрения для использования в различных программах межлабораторного контроля качества, улучшая тем самым эффективность проведения РНС в будущем. Нами было показано, что наборы Неоскрин имеют лучшую воспроизводимость и стабильность, а наборы TK-СМА – больший процент подтвержденных на ретесте образцов среди обследованных, выданных в группу риска.
При проведении РНС летальный исход в ряде случаев может наступать еще до отправки биоматериала на подтверждающую диагностику. Возможность проведения подтверждающей биохимической и молекулярно-генетической диагностики с использованием имеющегося биоматериала для семей таких пациентов играет особенно важную роль. С сожалением стоит отметить, что тяжелые формы болезни, приводящие к раннему летальному исходу, как в случае описанных нами детей с глутаровой ацидурией, тип 2С, и дефицитом митохондриального трифункционального белка, порой невозможно выявить даже в сжатые сроки, установленные нормативными документами РНС. Как и для других НБО интоксикационного типа, для этих нозологий характерен клинический полиморфизм, обусловленный тяжестью дефицита фермента. При этом симптомы заболевания могут проявиться в любом возрасте, но наиболее опасным в плане неблагоприятного исхода болезни является неонатальный период. Для указанных выше патологий существуют эффективные методы лечения, начиная от диетотерапии и заканчивая трансплантацией печени. Терапия дефицита митохондриального трифункционального белка сходна с таковой у пациентов с нарушением митохондриального -окисления жирных кислот с длинной и очень длинной углеродной цепью и включает в себя использование специализированных продуктов лечебного питания с модифицированным жировым компонентом. При этом значимость быстрой молекулярно-генетической верификации диагноза для таких пациентов особенно важна, поскольку позволяет в дальнейшем проводить медико-генетическое консультирование, предупреждая повторные случаи рождения детей с заболеванием в отягощенных семьях.
«Случайные находки» заслуживают отдельного внимания и также требуют повышенной настороженности специалистов широкого профиля. Так, например, в случае ультраредкого заболевания – этилмалоновой энцефалопатии, не включенной в перечень заболеваний РНС, спасти ребенка не удалось, и молекулярно-генетическое исследование было выполнено нами уже после смерти ребенка. Частота заболевания в РФ не установлена, а в мире к настоящему моменту описано около 100 случаев [46]. Заболевание не имеет специфической патогенетической терапии, характеризуется тяжелым прогрессирующим течением и требует мультидисциплинарного подхода таких специалистов, как невролог, гастроэнтеролог, генетик и др. Прогноз без поддерживающей терапии неблагоприятный, а исход напрямую зависит от тяжести клинических проявлений. Такие примеры доказывают безусловную значимость проведения подтверждающей молекулярно-генетической диагностики, а также требуют особого внимания к анализу лабораторных показателей, не включенных в настоящий момент в программу РНС.
В трех случаях дефицита 3-метилкротонил-КоА-карбоксилазы, который также не входит в программу РНС, возможно применение диетотерапии для контроля состояния детей. Дефицит 3-метилкротонил-КоА-карбоксилазы (3-MCCD) – это аутосомно-рецессивный врожденный дефект катаболизма лейцина, вызванный патогенными вариантами генов MCCC1 или MCCC2. Состояние характеризуется относительно доброкачественным течением и благоприятным исходом, однако, в некоторых случаях может понадобиться низкобелковая диета с использованием специализированного продукта лечебного питания без лейцина (назначается также при изовалериановой ацидурии), а также назначение карнитина.
Еще один важный аспект – это сложные диагностические случаи, требующие опыта и возможности проведения различных молекулярно-генетических исследований для верификации диагноза. Так, у одного из детей с подозрением на ГФА в результате подтверждающей молекулярно-генетической диагностики методом высокопроизводительного секвенирования в Центре 3Б был обнаружен лишь гетерозиготный вариант в гене PTS. При поступлении ребенка в наш центр была продолжена молекулярно-генетическая диагностика, в результате которой выявлен второй каузальный вариант – дупликация экзона 2 гена PTS chr11:112228590-112230231dup в гетерозиготном состоянии, что позволило подтвердить диагноз и инициировать лечение ребенка. Для лечения ГФА, обусловленной патогенными вариантами гена PTS, необходим пожизненный прием сапроптерина дигидрохлорида, синтетического аналога ВН4 (тетрагидробиоптерина). Препарат зарегистрирован в России, входит в список ЖНВЛП, включен в клинические рекомендации.
Случаи редких болезней, выявленные у детей, которым по различным причинам не был проведен РНС, либо в рамках проведения РНС были получены ложноотрицательные результаты, требуют особой настороженности и слаженной работы как специалистов центров 3А, так и медицинских работников, в том числе и первичного звена в регионах. Как мы видим из приведенных примеров, поздняя диагностика может приводить к необратимым и жизнеугрожающим последствиям для их здоровья. Так, например, поздняя диагностика у ребенка глутаровой ацидурии, тип 1, и, как следствие, запоздалое назначение терапии привели к его глубокой инвалидизации и паллиативному статусу.
В случае пациента со СМА, имеющего 2 копии гена SMN2, необследованного в рамках РНС в установленные сроки и выявленного нами по программе селективного скрининга в возрасте 6-ти месяцев, в результате слаженной работы генетиков, неврологов и представителей фонда «Круг добра» была инициирована генотерапия.
В контексте вышесказанного, особую значимость приобретает работа региональных специалистов, разъясняющая родителями и иным законными представителям новорожденных исключительную важность как этапа подтверждающей диагностики, так и проведения РНС в целом.
Неотъемлемой частью комплекса терапевтических мероприятий при ведении пациентов с редкими болезнями, выявленными в рамках РНС, является совместная работа членов семьи и специалистов лечебного учреждения, где наблюдаются дети с установленными заболеваниями, предопределяя эффективность назначенной терапии и снижая риск экстренной госпитализации.
Заключение
Реализация федеральной программы РНС, стартовавшей 01 января 2023 года, является сложным методическим процессом, направленным на раннее, досимптоматическое выявление новорожденных с орфанными заболеваниями. Представленные результаты скрининга 324 734 новорожденных из 22 субъектов ЦФО и ДФО показали высокую эффективность программы: охват скринингом составил 97,6%, частота выявленных случаев составила 1:1510 новорожденных, под динамическое наблюдение поставлено 93,5% детей с выявленными орфанными болезнями. Оптимизация алгоритма молекулярно-генетической диагностики ПИД, основанная на аналитических характеристиках наборов, позволила увеличить эффективность скрининга. Высокие частоты отдельных НБО, таких как ГФА (1:3865) и дефицит среднецепочечной ацил-КоА-дегидрогеназы жирных кислот (1:21648), а также мажорные варианты генов PAH и ACADM, выявленные в ходе работы, имеют важное практическое значение и могут быть использованы для внедрения программ профилактики в отдельных субъектах РФ.
_____________________________________________________________________
1 Федеральный закон от 21.11.2011 № 323-ФЗ «Об основах охраны здоровья граждан в Российской Федерации». URL: https://minzdrav.gov.ru/documents/7025-federalnyy-zakon-323-fz-ot-21-noyabrya-2011-g.
2 Распоряжение Правительства РФ от 09.06.2022 № 1510-р «О закупке и распределении ФКУ «Федеральный центр планирования и организации лекарственного обеспечения граждан» Министерства здравоохранения РФ медицинского оборудования для осуществления расширенного неонатального скрининга» (с изменениями и дополнениями). URL: https://base.garant.ru/404819443/?ysclid=m7c27si46o519351152.
3 Приказ Минздрава России от 27.12.2022 № 808н «Об утверждении перечней федеральных государственных медицинских организаций и государственных медицинских организаций, относящихся к ведению субъектов Российской Федерации, осуществляющих расширенный неонатальный скрининг, а также осуществляющих проведение подтверждающей биохимической, и (или) молекулярно-генетической, и (или) молекулярно-цитогенетической диагностики, и прикрепленных к ним субъектов Российской Федерации». URL: https://www.garant.ru/products/ipo/prime/doc/405966317/?ysclid=m7c2ac357p920558923.
4 Наименования торговых марок реактивов, систем для автоматического выделения и очистки нуклеиновых кислот, детектирующих амплификаторов, масс-спектрометров, программного обеспечения в публикации не приводятся. За уточнением торговых марок оборудования, используемого в исследовании, просьба обращаться к авторам или в редакцию.
- Chung C.C.Y., Chu A.T.W., Chung B.H.Y. Rare disease emerging as a global public health priority // Front Public Health. 2022. Vol. 10.
- European Medicines Agency (EMA) [Electronic resource]. URL: https://www.ema.europa.eu/en/homepage (accessed: 12.02.2025).
- Haendel M. [et al.] How many rare diseases are there? // Nat Rev Drug Discov. 2020. Vol. 19, № 2. P. 77–78.
- Home – EURORDIS [Electronic resource]. URL: https://www.eurordis.org/ (accessed: 12.02.2025).
- Orphan medicines figures 2000-2023 [Electronic resource] // https://efim.org/node/141259.
- Voronin S.V., Kutsev S.I. Neonatal screening for hereditary diseases in Russia: yesterday, today, and tomorrow // Neonatology: News, Opinions, Training. 2022. Vol. 10, № 4. P. 34–39.
- Wilson J.M., Jungner Y.G. Principles and practice of mass screening for disease. // Bol Oficina Sanit Panam. 1968. Vol 65, № 4. P. 281–393.
- Gonzalez J., Willis M.S. Robert Guthrie, MD, PhD // Lab Med. 2009. Vol. 40, № 12. P. 748–749.
- Arnold G.L. Inborn errors of metabolism in the 21st century: past to present // Ann Transl Med. 2018. Vol. 6, № 24. P. 467–467.
- Fonseca T., Macedo M.F. Inherited Metabolic Disorders: From Bench to Bedside // Biomedicines. 2024. Vol. 12, № 1. P. 174.
- Tangye S.G. et al. Human Inborn Errors of Immunity: 2022 Update on the Classification from the International Union of Immunological Societies Expert Committee // J Clin Immunol. 2022. Vol. 42, № 7. P. 1473–1507.
- van der Burg M. [et al.] Universal Newborn Screening for Severe Combined Immunodeficiency (SCID) // Front Pediatr. 2019. Vol. 7.
- Routes J.M. Statewide Newborn Screening for Severe T-Cell Lymphopenia // JAMA. 2009. Vol. 302, № 22. P. 2465.
- Barbaro M. [et al.] Newborn Screening for Severe Primary Immunodeficiency Diseases in Sweden a 2-Year Pilot TREC and KREC Screening Study // J Clin Immunol. 2017. Vol. 37, № 1. P. 51–60.
- van Zelm M.C. [et al.] Replication history of B lymphocytes reveals homeostatic proliferation and extensive antigen-induced B cell expansion // J Exp Med. 2007. Vol. 204, № 3. P. 645–655.
- Nakagawa N. [et al.] Quantification of -deleting recombination excision circles in Guthrie cards for the identification of early B-cell maturation defects // Journal of Allergy and Clinical Immunology. 2011. Vol. 128, № 1. P. 223-225.e2.
- Leontyeva M.E. [et al.] Extended Neonatal Screening for the Immune System Congenital Defects Detection: Experience of the Moscow Center for Neonatal Screening // Pediatria. Journal named after G.N. Speransky. 2024. Vol. 103, № 2. P. 44–51.
- Voronin S.V. [et al.] Advanced neonatal screening for hereditary diseases in Russia: first results and future prospects // Pediatria. Journal named after G.N. Speransky. 2024. Vol. 103, № 1. P. 16–29.
- Therrell B.L. [et al.] Current Status of Newborn Bloodspot Screening Worldwide 2024: A Comprehensive Review of Recent Activities (2020–2023) // Int J Neonatal Screen. 2024. Vol. 10, № 2. P. 38.
- White S. et al. Expanding the Australian Newborn Blood Spot Screening Program using genomic sequencing: do we want it and are we ready? // European Journal of Human Genetics. 2023. Vol. 31, № 6. P. 703–711.
- Dosi C., Masson R. The impact of three SMN2 gene copies on clinical characteristics and effect of disease-modifying treatment in patients with spinal muscular atrophy: a systematic literature review // Front Neurol. 2024. Vol. 15.
- Куцев С.И. Неонатальный скрининг. Национальное руководство. 2023.
- Spiekerkotter U., Krude H. Target diseases for neonatal screening in Germany—challenges for treatment and long-term care // Dtsch Arztebl Int. 2022.
- Mutze U. [et al.] Sudden neonatal death in individuals with medium-chain acyl-coenzyme A dehydrogenase deficiency: limit of newborn screening // Eur J Pediatr. 2022. Vol. 181, № 6. P. 2415–2422.
- Pronina N. [et al.] The molecular basis of phenylketonuria in Latvia // Hum Mutat. 2003. Vol. 21, № 4. P. 398–399.
- Zschocke J. Phenylketonuria mutations in Europe // Hum Mutat. 2003. Vol. 21, № 4. P. 345–356.
- Kuypers A.M. [et al.] Evaluation of Neonatal Screening Programs for Tyrosinemia Type 1 Worldwide // Int J Neonatal Screen. 2024. Vol. 10, № 4. P. 82.
- Sander J. [et al.] Neonatal screening for citrullinaemia // Eur J Pediatr. 2003. Vol. 162, № 6. P. 417–420.
- Rohit P., Dey S., Tank T. Maple Syrup Urine Disease : A Rare Inherted Disorder of Amino Acid Metabolism // IJFMR240426452. 2024. Vol. 6, № 4.
- Lindner M. [et al.] Neonatal screening for glutaric aciduria type I: Strategies to proceed // J Inherit Metab Dis. 2006. Vol. 29, № 2–3. P. 378–382.
- Elola Pastor A.I., Prieto Garc a B., D az Mart n J.J. Evaluation of the first 5 years of a glutaric aciduria type I neonatal screening programme in Asturias // Anales de Pediatr a (English Edition). 2024. Vol. 100, № 5. P. 318–324.
- Boy N. [et al.] Newborn screening: A disease-changing intervention for glutaric aciduria type 1 // Ann Neurol. 2018. Vol. 83, № 5. P. 970–979.
- Dangouloff T. [et al.] Newborn screening programs for spinal muscular atrophy worldwide: Where we stand and where to go // Neuromuscular Disorders. 2021. Vol. 31, № 6. P. 574–582.
- Hale K., Ojodu J., Singh S. Landscape of Spinal Muscular Atrophy Newborn Screening in the United States: 2018–2021 // Int J Neonatal Screen. 2021. Vol. 7, № 3. P. 33.
- Efimova I.Yu. [et al.] Epidemiology of Spinal Muscular Atrophy Based on the Results of a Large-Scale Pilot Project on 202,908 Newborns // Pediatr Neurol. 2024. Vol. 156. P. 147–154.
- Muller-Felber W. [et al.] Infants Diagnosed with Spinal Muscular Atrophy and 4 SMN2 Copies through Newborn Screening – Opportunity or Burden? // J Neuromuscul Dis. 2020. Vol. 7, № 2. P. 109–117.
- Wadman R.I. [et al.] Intragenic and structural variation in the SMN locus and clinical variability in spinal muscular atrophy // Brain Commun. 2020. Vol. 2, № 2.
- Niba E.T.E. [et al.] Clinical phenotypes of spinal muscular atrophy patients with hybrid SMN gene // Brain Dev. 2021. Vol 43, № 2. P. 294–302.
- Costa-Roger M. [et al.] The Importance of Digging into the Genetics of SMN Genes in the Therapeutic Scenario of Spinal Muscular Atrophy // Int J Mol Sci. 2021. Vol. 22, № 16. P. 9029.
- Sumner C.J., Crawford T.O. Early treatment is a lifeline for infants with SMA // Nat Med. 2022. Vol. 28, № 7. P. 1348–1349.
- Notarangelo L.D., Uzel G., Rao V.K. Primary immunodeficiencies: novel genes and unusual presentations // Hematology. 2019. Vol. 2019, № 1. P. 443–448.
- Rechavi E. [et al.] Newborn Screening for Severe Combined Immunodeficiency in Israel // Int J Neonatal Screen. 2017. Vol. 3, № 2. P. 13.
- Strand J. [et al.] Second-Tier Next Generation Sequencing Integrated in Nationwide Newborn Screening Provides Rapid Molecular Diagnostics of Severe Combined Immunodeficiency // Front Immunol. 2020. Vol. 11.
- Argudo-Ram rez A. [et al.] First Universal Newborn Screening Program for Severe Combined Immunodeficiency in Europe. Two-Years’ Experience in Catalonia (Spain) // Front Immunol. 2019. Vol. 10.
- Lev A., Somech R., Somekh I. Newborn screening for severe combined immunodeficiency and inborn errors of immunity // Curr Opin Pediatr. 2023. Vol. 35, № 6. P. 692–702.
- Dionisi-Vici C. [et al.] Liver transplant in ethylmalonic encephalopathy: a new treatment for an otherwise fatal disease // Brain. 2016. Vol. 139, № 4. P. 1045–1051.
- Савостьянов К.В. Современные алгоритмы генетической диагностики редких наследственных болезней у российских пациентов : Монография / К.В. Савостьянов. – Москва: ООО «Полиграфист и издатель», 2022. – 452 с. – ISBN 978-5-6047928-7-2. – EDN RDUZGH.