КОНТРОЛЬ КАЧЕСТВА И БЕЗОПАСНОСТИ
Общие требования безопасности и эффективности медицинских изделий как основа формирования регистрационного досье медицинского изделия в целях его регистрации в рамках Евразийского экономического союза
Информация об авторах
1 — Федеральная служба по надзору в сфере здравоохранения, Российская Федерация, 109074, Москва, Славянская площадь, д. 4, стр. 1.
2 — Федеральная служба по надзору в сфере здравоохранения, Российская Федерация, 109074, Москва, Славянская площадь, д. 4, стр. 1.
Опубликовано: 25.06.2021
Статья посвящена вопросам, связанным с подходами к формированию регистрационного досье медицинского изделия в целях его регистрации в рамках Евразийского экономического союза. В частности, обосновывается важность заполнения документа «Сведения о соответствии медицинского изделия общим требованиям безопасности и эффективности медицинских изделий, требованиям к их маркировке и эксплуатационной документации на них» с точки зрения подготовки документов регистрационного досье.
Ключевые слова: медицинские изделия, Евразийский экономический союз, регистрационное досье, Общие требования безопасности и эффективности медицинских изделий
Введение
Единые принципы обращения медицинских изделий в рамках Евразийского экономического союза (далее – Союз) определены в Соглашении о единых принципах и правилах обращения медицинских изделий (изделий медицинского назначения и медицинской техники) в рамках Евразийского экономического союза от 23.12.2014 (далее – Соглашение).
В соответствии с частью 2 статьи 3 Соглашения государства-члены Союза проводят скоординированную политику в сфере обращения медицинских изделий посредством:
- установления общих требований безопасности и эффективности медицинских изделий в рамках Союза;
- установления единых правил обращения медицинских изделий в соответствии с рекомендациями Международного форума регуляторов медицинских изделий (IMDRF);
- определения единых подходов к созданию системы обеспечения качества медицинских изделий.
В целях реализации положений Соглашения в настоящее время принято более 20 нормативных правовых актов, регламентирующих обращение медицинских изделий в рамках Союза.
Хотелось бы отметить, что в соответствии с Соглашением регулирование в сфере медицинских изделий носит «отсылочный» характер, то есть на законодательном уровне принимаются базовые требования в отношении безопасности продукции, сформулированные в настоящее время в Общих требованиях безопасности и эффективности медицинских изделий, требованиях к их маркировке и эксплуатационной документации на них, утвержденных решением Совета Евразийской экономической комиссии от 12.02.2016 № 27 (далее – Общие требования), а детальные требования безопасности раскрываются в выбранных («признанных») стандартах, которые обеспечивают презумпцию соответствия Общим требованиям. [1]
Основные группы документов регистрационного досье на медицинское изделие
Перечень документов, представляемых в целях регистрации медицинского изделия в рамках Союза, представлен в приложении 4 к Правилам регистрации и экспертизы безопасности, качества и эффективности медицинских изделий, утвержденным решением Совета Евразийской экономической комиссии от 12.02.2016 № 46 (далее – Правила). При анализе данного перечня все документы досье можно разделить на несколько групп (табл. 1).
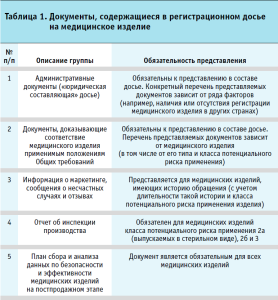
Необходимо отметить, что самой большой группой документов (около 60% документов, перечисленных в приложении 4 к Правилам) является группа, содержащая доказательства соответствия медицинского изделия применимым положениям Общих требований. Таким образом, основной целью при подготовке досье заявителем должен являться сбор документов, материалов и сведений, обеспечивающих доказательство соответствия регистрируемого медицинского изделия Общим требованиям. В связи с этим возникает вопрос, что же представляют собой Общие требования и какие существуют способы доказательства соответствия им.
Способы доказательства соответствия регистрируемого медицинского изделия Общим требованиям
Общие требования являются документом, гармонизированным с двумя международными документами:
- в части требований безопасности и эффективности действующая редакция Общих требований гармонизирована с международным документом GHTF/ SG1/N68:2012 Essential Principles of Safety and Performance of Medical Devices. Следует отметить, что в октябре 2018 г. принята обновленная версия указанного документа: IMDRF GRRP WG/N47 Essential Principles of Safety and Performance of Medical Devices and IVD Medical Devices. Указанные «Основные принципы безопасности и эффективности медицинских изделий» являются одним из основополагающих документов в модели регулирования медицинских изделий, принятой в IMDRF. В нем содержится перечень основных критериев высокого уровня в течение всего жизненного цикла медицинских изделий, которые при их соблюдении дают гарантию того, что медицинские изделия безопасны и работают в соответствии с назначением, определенным производителем.
- в части требований к маркировке и сопроводительной документации Общие требования также гармонизированы с документом GHTF/SG1/N70:2011 Label and Instructions for Use for Medical Devices. Принятые в Союзе Общие требования состоят из пяти разделов.
Первым разделом являются «Общие положения», в которых приведены сведения о понятиях и определениях, используемых в документе.
Во втором разделе приведены «Общие требования безопасности и эффективности, применимые ко всем медицинским изделиям». Всего в данном разделе представлено девять таких требований, включающих в том числе:
- Требования к эффективности медицинских изделий. Примером таких требований является п. 6 Общих требований, согласно которому медицинские изделия должны быть эффективными так, как это предусмотрено производителем, и должны быть спроектированы и изготовлены таким образом, чтобы в нормальных условиях эксплуатации они соответствовали целям применения по назначению, определенному производителем.
- Общие требования безопасности. Следует отметить, что в соответствии с п. 4 Общих требований решения, принятые производителем при проектировании и изготовлении медицинского изделия, должны соответствовать принципам безопасности с учетом общепризнанного уровня развития знаний.
- Концепцию минимального риска. В соответствии с п. 8 Общих требований все известные и предполагаемые риски, возникающие при использовании медицинского изделия, и любые нежелательные эффекты от такого использования сводятся к минимуму и должны быть приемлемыми при сопоставлении с пользой для пользователей, получаемой от предусмотренного производителем действия медицинского изделия при нормальных условиях эксплуатации.
- Общие требования к предоставляемой потребителю информации, в том числе информирование об остаточных рисках.
Следует отметить, что в соответствии с ГОСТ ISO 14971-2011 «Изделия медицинские. Применение менеджмента риска к медицинским изделиям» в отношении риска, где польза от применения медицинского изделия превышает риск, изготовитель должен решить, какую информацию по безопасности необходимо предоставить для информирования об остаточном риске.
Относительно требований, указанных в данном разделе, следует отметить, что:
- рекомендации по анализу и оценке доказательных материалов (документов), представляемых производителем с целью подтверждения соответствия указанным требованиям, представлены в Методических рекомендациях по проведению экспертизы безопасности, качества и эффективности медицинских изделий в целях их регистрации в рамках Евразийского экономического союза, утвержденных рекомендациях Коллегии Евразийской экономической комиссии от 21.05.2019 № 14 (далее – Методические рекомендации). Данные рекомендации могут быть полезны заявителю при сборе доказательных материалов;
- в соответствии с п. 112 Общих требований доказательства соответствия медицинского изделия положениям, установленным пунктами 3, 6 и 8 Общих требований, должны включать клиническое обоснование на основе клинических данных о медицинском изделии.
Третий и четвертый раздел Общих требований посвящен «Общим требованиям безопасности и эффективности, применимым к медицинским изделиям, за исключением медицинских изделий для диагностики in vitro», а также «Общим требованиям безопасности и эффективности, применяемым к медицинским изделиям для диагностики in vitro». Относительно данных разделов необходимо отметить следующее:
- данные разделы содержат частные требования безопасности и эффективности, например, в части защиты от излучения, инфекционного и микробного загрязнения медицинских изделий и т.д. Таким образом, применимость данных требований зависит от конкретного медицинского изделия;
- многие из перечисленных в данных разделах требований основаны на концепции минимального риска;
- в каждом из указанных разделов имеются подразделы, регламентирующие требования к маркировке медицинских изделий и требования к информации, содержащейся в инструкции по применению медицинского изделия.
Последним, но при этом не менее важным, является пятый раздел, посвященный доказательствам соответствия медицинских изделий общим требованиям безопасности и эффективности в целях регистрации.
В соответствии с п. 111 Общих требований сведения о соответствии медицинского изделия Общим требованиям представляются производителем в виде таблицы, заполненной по форме, представленной в таблице 2.

В соответствии с приложением 4 к Правилам, данная таблица является отдельным документом досье, называемым «Сведения о соответствии медицинского изделия общим требованиям безопасности и эффективности медицинских изделий, требованиям к их маркировке и эксплуатационной документации на них». Отмечаем, что данный документ является одним из самых важных документов в досье.
В связи с этим рассмотрим основные подходы к оформлению «Сведений о соответствии медицинского изделия общим требованиям безопасности и эффективности медицинских изделий, требованиям к их маркировке и эксплуатационной документации на них», а также укажем основные ошибки, выявляемые при рассмотрении досье, поступающих в Росздравнадзор.
В графе 3 таблицы 2 указывается метод, примененный для доказательства соответствия медицинского изделия требованию, указанному в графе 1.
В соответствии с п. 109 Общих требований соответствие медицинского изделия Общим требованиям может обеспечиваться:
- Выполнением требований соответствующих разделов стандартов, включенных в Перечень стандартов, в результате применения которых на добровольной основе полностью или частично обеспечивается соблюдение соответствия медицинских изделий Общим требованиям безопасности и эффективности медицинских изделий, требованиям к их маркировке и эксплуатационной документации на них, утвержденный рекомендацией Коллегии Евразийской экономической комиссии от 04.09.2017 № 17 (далее – Перечень).
Следует отметить, что подтверждение соответствия требованиям применимых к медицинскому изделию стандартов, включенных в Перечень, является гарантированным способом («презумпцией соответствия») доказательства соответствия Общим требованиям.
При этом:
- если для доказательства соответствия медицинского изделия Общим требованиям производитель применяет не все структурные элементы стандарта, предусмотренные Перечнем, то он должен представить в составе досье обоснование их неприменения, доказывающее, что сделанные изъятия не снижают безопасность и (или) эффективность рассматриваемого медицинского изделия;
- если производитель применяет более позднюю версию стандарта, включенного в Перечень, то презумпция соответствия медицинского изделия Общим требованиям распространяется только на те нормы и требования применяемого стандарта, которые не изменяют или не снижают требования к медицинскому изделию по отношению к требованиям стандарта, включенного в Перечень. Для подтверждения того, что нормы и требования применяемого стандарта не снижают требования к медицинскому изделию по отношению к требованиям стандарта, включенного в Перечень, заявитель представляет таблицу изменений для применяемых структурных элементов стандарта с анализом имеющихся различий.
Подтверждение соответствия медицинского изделия стандарту, включенному в Перечень, может быть выполнено следующими способами:
а) доказательствами уполномоченных организаций, то есть проведением испытаний (исследований) в соответствии с Правилами проведения технических испытаний медицинских изделий, утвержденными решением Совета Евразийской экономической комиссии от 12.02.2016 № 28, или Правилами проведения исследований (испытаний) с целью оценки биологического действия медицинских изделий, утвержденными решением Совета Евразийской экономической комиссии от 16.05.2016 № 38, в выбранных заявителем учреждениях и организациях, имеющих право проводить испытания (исследования) в целях регистрации медицинских изделий и включенных в единый реестр уполномоченных организаций Союза1.
При этом испытания (исследования) на соответствие требованиям стандартов, включенных в Перечень, являющихся стандартами на продукцию, проводятся уполномоченной организацией с применением методов испытаний (исследований), содержащихся в соответствующих включенных в Перечень стандартах;
б) доказательствами первой стороны в случаях, когда соответствующий стандарт, включенный в Перечень, применяется к медицинским изделиям, однако отсутствует стандартизованный или аттестованный в установленном порядке метод испытания (исследования).
Обращаем внимание, что в соответствии с Методическими рекомендациями доказательствами первой стороны являются собственные доказательные материалы (документы) производителя и (или) уполномоченного представителя производителя (протоколы собственных испытаний (исследований), в том числе выданные органами по сертификации, испытательными лабораториями (центрами), аккредитованными в национальной системе аккредитации государства – члена Союза, результаты клинического применения и другие материалы).
Хотелось бы отметить, что в данном случае в досье необходимо представить обоснование относительно валидности применяемого метода испытания (исследования) для подтверждения соответствия стандарту, включенному в Перечень, с учетом класса потенциального риска применения медицинского изделия.
- Выполнением установленных этим документом требований непосредственно. Данный способ подразумевает представление заявителем доказательных материалов (документов), которые могут включать в себя доказательства первой стороны, доказательства уполномоченных организаций, в том числе результаты испытаний (исследований) на соответствие требованиям применяемых стандартов, не включенных в Перечень.
Хотелось бы отметить, что производитель вправе применять стандарты и методы, не включенные в Перечень, однако их применение не обеспечивает презумпцию соответствия медицинского изделия Общим требованиям. В связи с этим производитель представляет обоснование валидности применяемого стандарта и (или) метода для подтверждения соответствия медицинского изделия Общим требованиям.
В графе 4 таблицы 2 указываются реквизиты нормативного документа на метод, используемый для доказательства соответствия медицинского изделия требованию, указанному в графе 1.
Основными ошибками при заполнении данной графы являются:
- указание не в полном объеме реквизитов нормативного документа на метод, используемый для доказательства соответствия медицинского изделия тому или иному пункту Общих требований;
- отсутствие конкретизации применяемых структурных элементов стандарта (в случае если производитель в целях доказательства соответствия медицинского изделия Общим требованиям применяет не все структурные элементы стандарта).
В графе 5 таблицы 2 указываются реквизиты документов, подтверждающих соответствие медицинского изделия требованию, указанному в графе 1. Такими документами могут являться, например, протоколы испытаний, сертификаты, декларации соответствия, отчеты о проведенных исследованиях и другие документы.
Следует отметить, что все документы, ссылки на которые указаны в данной графе, должны быть представлены в составе досье на медицинское изделие вне зависимости от того, перечислены ли они в приложении 4 к Правилам или нет. Данное требование связано с тем, что именно на основании анализа документов, перечисленных в графе 5 таблицы 2, делается вывод о безопасности и эффективности регистрируемого изделия.
Основной ошибкой при заполнении данной графы является отсутствие конкретизации сведений о реквизитах документов (наименовании документа, его дате и регистрационном номере) и указание только их наименований. Обращаем внимание, что законодательством Союза предусмотрена обязанность поддержания ряда документов досье, например, отчета о клиническом доказательстве эффективности и безопасности медицинского изделия, в актуальном состоянии. В связи с этим перед подачей досье в уполномоченный орган (экспертную организацию) референтного государства необходимо проверить актуальность предоставляемых документов.
Заключение
В завершении следует отметить, что правильно заполненный документ «Сведения о соответствии медицинского изделия общим требованиям безопасности и эффективности медицинских изделий, требованиям к их маркировке и эксплуатационной документации на них» может стать для заявителя «маршрутной картой» при сборе документов, значительно упростить процесс подготовки регистрационного досье и стать гарантом успешной регистрации медицинского изделия в рамках Союза.
_______________________________________________________________________
1 Реестр уполномоченных организаций размещается в открытой части информационной системы Союза в сфере обращения медицинских изделий на информационном портале Союза в информационно-телекоммуникационной сети «Интернет». Национальная часть реестра уполномоченных организаций дополнительно размещена на официальном сайте Росздравнадзора в разделе «Медицинские изделия» «Регистрация медицинских изделий в рамках Евразийского экономического союза».
1. Антонов В.С. Общие требования безопасности и эффективности медицинских изделий в модели регулирования Евразийского экономического союза. // Вестник Росздравнадзора. – 2016. – № 5. – С. 14–16.