МЕДИЦИНСКИЕ ИЗДЕЛИЯ
Клиническая апробация как мост между наукой и практикой: десятилетний опыт и перспективы развития
Информация об авторах
1 — ФГБУ «Национальный медицинский исследовательский центр травматологии и ортопедии имени Р.Р. Вредена» Министерства здравоохранения Российской Федерации, 195427, Российская Федерация, г. Санкт-Петербург, ул. Академика Байкова, д. 8.
2 — ФГБУ «Национальный медицинский исследовательский центр травматологии и ортопедии имени Р.Р. Вредена» Министерства здравоохранения Российской Федерации, 195427, Российская Федерация, г. Санкт-Петербург, ул. Академика Байкова, д. 8.
3 — ФГБУ «Национальный медицинский исследовательский центр травматологии и ортопедии имени Р.Р. Вредена» Министерства здравоохранения Российской Федерации, 195427, Российская Федерация, г. Санкт-Петербург, ул. Академика Байкова, д. 8.
4 — ФГБУ «Центр экспертизы и контроля качества медицинской помощи» Минздрава России, 109028, Российская Федерация, г. Москва, Покровский б-р, д. 6/20 стр. 2.
5 — ФГБУ «Национальный медицинский исследовательский центр травматологии и ортопедии имени Р.Р. Вредена» Министерства здравоохранения Российской Федерации, 195427, Российская Федерация, г. Санкт-Петербург, ул. Академика Байкова, д. 8.
6 — ФГБУ «Национальный медицинский исследовательский центр травматологии и ортопедии имени Р.Р. Вредена» Министерства здравоохранения Российской Федерации, 195427, Российская Федерация, г. Санкт-Петербург, ул. Академика Байкова, д. 8.
7 — ФГБУ «Национальный медицинский исследовательский центр травматологии и ортопедии имени Р.Р. Вредена» Министерства здравоохранения Российской Федерации, 195427, Российская Федерация, г. Санкт-Петербург, ул. Академика Байкова, д. 8.
Опубликовано: 27.04.2026
В современной травматологии и ортопедии ревизионное эндопротезирование тазобедренного сустава при обширных костных дефектах остается одной из наиболее сложных клинических ситуаций. Частота выявления дефектов 3А и 3B типов по классификации W. Paprosky среди пациентов, направляемых на повторные оперативные вмешательства, достигает высоких процентов в специализированных центрах. Применение стандартных серийных ревизионных систем в таких условиях нередко не обеспечивает первичной механической стабильности, что закономерно приводит к повышенному риску асептического расшатывания, переломов имплантата, инфекционных осложнений и, как следствие, повторных ревизий. [1–7].
Перспективной альтернативой, активно развивающейся в последнее десятилетие, стали персонализированные имплантаты, изготавливаемые методом аддитивных технологий (3D-печать) на основе данных компьютерной томографии конкретного пациента. Такие конструкции позволяют с высокой анатомической точностью воспроизводить форму и размеры дефекта, обеспечивать оптимальную первичную стабильность, стимулировать остеоинтеграцию за счет пористых структур и достигать функционально приемлемого результата в ситуациях, где традиционные методы оказываются недостаточно эффективными. [8, 9]
Ключевые слова: отечественные медицинские изделия, травматология, ортопедия, ревизионное эндопротезирование тазобедренного сустава, персонализированный имплантат, аддитивные технологии, инновационные технологии
Введение
Развитие направления дорогостоящего персонализированного эндопротезирования в Российской Федерации стало возможным благодаря внедрению в практику протоколов клинической апробации, разработанных в НМИЦ травматологии и ортопедии им. Р.Р. Вредена при методической поддержке ФГБУ «Центр экспертизы и контроля качества медицинской помощи» Минздрава России и утвержденных Министерством здравоохранения Российской Федерации. Клиническая апробация представляет собой регламентированную процедуру оценки эффективности и безопасности новых методов лечения в условиях ограниченной выборки пациентов при строгом этическом контроле, вместе с тем позволяющей обеспечить достоверность получаемых результатов. Ее ключевая цель – создание доказательной базы, достаточной для последующего включения метода в клинические рекомендации и систему государственных гарантий.
Знаковым является тот факт, что в прошлом году исполнилось 10 лет с момента запуска этого механизма в отечественном здравоохранении. За десятилетие клиническая апробация доказала свою состоятельность как эффективный инструмент «быстрого» перехода от лабораторного образца или научной гипотезы к реальной медицинской помощи.
Первый российский опыт: от единичной операции до системного подхода
На базе Национального медицинского исследовательского центра травматологии и ортопедии им. Р.Р. Вредена в 2015 году была выполнена первая в России операция с имплантацией индивидуального вертлужного компонента, изготовленного методом 3D-печати. Пациент имел выраженный посттравматический дефект костной ткани (3B по классификации W. Paprosky) со сложной деформацией вертлужной впадины в условиях неудачного первичного эндопротезирования.
Индивидуальный имплантат был спроектирован с учетом уникальной анатомии дефекта и напечатан из титанового сплава методом селективного лазерного плавления. (рис.1)
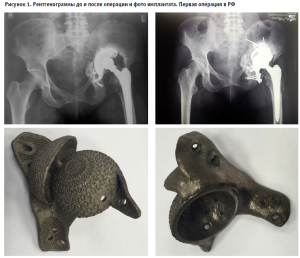
Уже в 2016 году был инициирован и утвержден первый профильный протокол клинической апробации, который четко определил показания к применению метода: наличие выраженных костных дефектов, невозможность использования стандартных ревизионных систем, ожидаемая эффективность от применения индивидуальной конструкции по данным предоперационного компьютерного моделирования (рис. 2) Протокол также регламентировал критерии исключения (активный инфекционный процесс, некомпенсированная соматическая патология, отказ пациента). Ключевое обстоятельство, позволившее столь быстро начать клиническое применение, заключается в юридическом статусе изделия: поскольку имплантат изготавливался индивидуально для каждого пациента и к нему предъявлялись специальные требования в соответствии с назначением, выданным медицинским работником, он подпадал под норму Соглашения о единых принципах и правилах обращения медицинских изделий (изделий медицинского назначения и медицинской техники) в рамках Евразийского экономического союза и не требовал регистрации уполномоченным органом. [10].

Это дало возможность проводить операции уже в 2015 году, минуя длительные регистрационные процедуры, исключительно на основании разрешения этического комитета в рамках утвержденного протокола апробации.
Организационная кооперация и технологическая цепочка
В рамках апробации была выстроена эффективная кооперация между медицинским учреждением и отечественными биоинжиниринговыми компаниями, обладающими мощностями для аддитивного производства. Отработана полная технологическая цепочка: проведение КТ-исследования с тонкими срезами, сегментация костных структур, 3D-моделирование дефекта и имплантата, инженерный анализ прочности, изготовление методом послойного лазерного спекания, финишная обработка, стерилизация и доставка в операционную. Важным достижением стало значительное сокращение среднего срока изготовления – с первоначальных 6–8 недель до 14–21 дня на текущий момент. Такое ускорение стало возможным благодаря созданию Центра компетенции с обязательным привлечением оперирующего хирурга и оптимизации процессов передачи данных, внедрению автоматизированных алгоритмов построения имплантата и использованию высокопроизводительных 3D-принтеров.
Результаты клинической апробации: безопасность и эффективность
Параллельно с накоплением клинического опыта проводились сравнительные исследования. В контрольную группу вошли пациенты, оперированные с применением стандартных ревизионных систем в том же центре. Группы были сопоставимы по возрасту, полу, индексу коморбидности и исходной степени дефекта.
Результаты подтвердили значительное снижение частоты послеоперационных осложнений при использовании персонализированных конструкций, а данные клинико-экономического анализа с использованием модели Маркова продемонстрировали, что, несмотря на более высокую стоимость индивидуального имплантата (в среднем на 40–50% дороже стандартного ревизионного набора), суммарные затраты на одного пациента при выраженном костном дефекте в горизонте трех лет оказались на 18–22% ниже за счет сокращения числа повторных ревизий, меньшей длительности стационарного лечения осложнений и более ранней реабилитации.[11]
Легитимизация метода: от апробации к клиническим рекомендациям и тарифу ОМС
Накопленный клинический опыт, подкрепленный данными наблюдения за более чем 200 пациентами, позволил включить метод в клинические рекомендации по коксартрозу в раздел «хирургическое лечение» – эндопротезирование в особо сложных случаях (утверждены в 2024 году) (рис. 3).
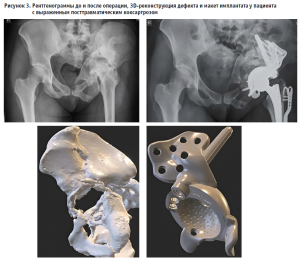
В 2025 году был утвержден новый вид высокотехнологичной медицинской помощи (профиль 16.00.80.002) «Ревизионное эндопротезирование суставов с использованием индивидуальных конструкций, изготовленных с применением аддитивных 3D-технологий» с установлением специализированного тарифа. Таким образом, клиническая апробация выполнила свою интегративную функцию: персонализированные аддитивные технологии легитимизированы и стали доступны широкому кругу пациентов на всей территории Российской Федерации. Юридическая конструкция самого механизма апробации зарекомендовала себя как полностью состоятельная для решения задач, связанных с индивидуальными медицинскими изделиями (рис. 4).

Серийные медицинские изделия: существующий порядок и неиспользованные возможности
Вместе с тем, анализируя десятилетний опыт применения клинической апробации, представляется целесообразным обратить внимание на смежную область – оценку серийных медицинских изделий, разрабатываемых в рамках научно-исследовательских работ в тех же научных клиниках. Под серийным изделием понимается конструкция, которая в перспективе может выпускаться стандартизированными партиями для неопределенного круга пациентов. Примерами являются оригинальная модель эндопротеза, пластины для остеосинтеза, интрамедуллярные фиксаторы с измененной геометрией.
В отличие от индивидуального имплантата, для вывода серийного изделия на рынок необходима государственная регистрация в Росздравнадзоре. При этом значительная часть серийных медицинских изделий регистрируется в Российской Федерации по варианту сравнения (биоэквивалентности или технической аналогии) без проведения клинических испытаний на человеке. Такая процедура допускается, если заявляемое изделие признается существенно эквивалентным уже зарегистрированному аналогу по техническим характеристикам, биосовместимости, предполагаемому применению и принципу действия. Регистрационное удостоверение выдается на основании результатов технических испытаний, токсикологических исследований, оценки эксплуатационной документации – но без единого случая имплантации человеку. И только после получения регистрационного удостоверения изделие впервые попадает в клиническую практику, что создает риск выявления нежелательных эффектов уже на этапе широкого применения.
Доклинические исследования – механические испытания на образцах (статическая и циклическая нагрузка, износ, коррозионная стойкость), эксперименты на животных, компьютерное моделирование методом конечных элементов – являются необходимой, но не всегда достаточной основой для прогнозирования поведения имплантата в организме человека. Различия в костном ремоделировании, иммунном ответе, характере реальных биомеханических нагрузок (особенно при движении с неконтролируемой амплитудой) и сроках остеоинтеграции могут приводить к тому, что результаты, полученные на животных моделях, не в полной мере воспроизводятся у человека.
В этой связи возникает вопрос: возможно ли создать дополнительный, факультативный этап оценки серийных изделий, который позволял бы до начала дорогостоящих регистрационных процедур получить первые данные о безопасности и предварительной эффективности на ограниченной группе добровольцев? Такой подход мог бы снизить риски как для пациентов (выявление потенциально неудачных конструкций до их широкого применения), так и для разработчиков (минимизация финансовых потерь в случае отрицательных результатов, возможность своевременной модификации изделия).
Действующие правовые ограничения и обоснование предложения
В настоящее время действующее законодательство не предусматривает для серийных медицинских изделий возможности проведения ограниченных клинических исследований на человеке после завершения доклинической фазы, но до подачи заявки на регистрацию. После создания опытных образцов в рамках научно-исследовательской работы в клинике, обладающей высоким хирургическим и научным потенциалом, исследователи не могут имплантировать эти образцы небольшой группе пациентов (5–10 человек) с разрешения этического комитета и ученого совета, поскольку для серийного изделия это требует регистрационного удостоверения. Единственный легальный путь – сразу выходить на полноценные клинические испытания в рамках регистрации, что предполагает значительные финансовые затраты и длительные сроки. На этапе опытного образца, когда еще отсутствует промышленный партнер и не подтверждена клиническая значимость конструкции, такие затраты могут быть трудновыполнимыми для академических коллективов. В результате ряд перспективных отечественных разработок не достигает стадии регистрации, оставаясь на уровне лабораторных прототипов, что замедляет технологическое развитие медицинской промышленности.
Предложение о введении дорегистрационных клинических испытаний (малой серии)
Учитывая успешный опыт клинической апробации индивидуальных изделий, представляется оправданным обсуждение возможности введения для серийных медицинских изделий понятия «дорегистрационные клинические испытания» (или клинические испытания малой серии). Такой инструмент мог бы быть реализован следующим образом:
- Условия проведения: научная медицинская организация, имеющая лицензию на медицинскую деятельность, аккредитованный этический комитет, опыт проведения НИР и необходимую материально-техническую базу (операционные, оборудование для послеоперационного наблюдения).
- Основание: положительные результаты доклинических исследований (механические, токсикологические, на животных), подтвержденные отчетами аккредитованных лабораторий.
- Объем: имплантация опытных образцов ограниченному числу пациентов (например, от 5 до 20) с целью оценки первичной безопасности (частота нежелательных явлений, отсутствие ранних миграций, реакций отторжения) и предварительной эффективности.
- Регулирование: проведение на базе одной клиники, финансирование за счет грантов, программ НИР или собственных средств разработчика, без требования регистрационного удостоверения. Достаточно разрешения этического комитета, ученого совета и информированного согласия пациента.
- Отличие от полноценных регистрационных испытаний: такие исследования не заменяют последующих исследований для регистрации. Их результат – научный отчет, который может служить обоснованием для принятия решения: либо запускать многоцентровые регистрационные испытания (если данные обнадеживающие), либо возвращаться к доработке конструкции (если выявлены существенные недостатки), либо прекратить разработку (при серьезных проблемах безопасности).
Потенциальные преимущества и нормативная реализация
Введение такого дополнительного этапа не потребовало бы кардинальной перестройки нормативной базы, а лишь дополнило бы ее, создав «мягкий» переход от доклинических исследований к полноценным регистрационным испытаниям. Это особенно актуально для изделий, разрабатываемых в академических и университетских клиниках, где инновационный потенциал высок, а финансовые ресурсы для масштабных регистрационных исследований ограничены. Кроме того, дорегистрационные испытания позволили бы на раннем этапе выявлять конструкции с неблагоприятным профилем безопасности, предотвращая их выход на рынок и потенциальный вред для пациентов. Также они могли бы способствовать развитию отечественного медицинского приборостроения, снижая барьеры для малых инновационных предприятий и стартапов.
Заключение
Резюмируя, следует отметить, что клиническая апробация, отметившая в 2025 году свое десятилетие, продемонстрировала высочайшую эффективность в качестве интегративного механизма, обеспечившего легитимизацию аддитивных технологий в отечественной травматологии и ортопедии. Персонализированные имплантаты, созданные методом 3D-печати, прошли путь от первой экспериментальной операции до утверждения отдельного вида высокотехнологичной медицинской помощи. Этот успех стал возможен благодаря гибкости правового режима, который позволил проводить клиническую оценку индивидуальных изделий без предварительной регистрации, под контролем этического комитета.
Вместе с тем, оглядываясь на десятилетний опыт, представляется целесообразным рассмотреть возможность распространения логики клинической апробации (ограниченное применение на небольшой группе пациентов под контролем этического комитета) на серийные медицинские изделия на этапе опытных образцов – в формате дорегистрационных клинических испытаний малой серии. Такое дополнение могло бы стать следующим шагом в развитии регуляторной среды, способствуя более плавному и безопасному переходу научных разработок в реальную клиническую практику, сохраняя при этом все завоевания существующей системы клинической апробации. Дальнейшее обсуждение этого предложения с участием представителей Росздравнадзора, научного сообщества и профильных ассоциаций может привести к выработке сбалансированного и юридически выверенного решения, которое ускорит внедрение отечественных инноваций без ущерба для безопасности пациентов.
- Ревизионное эндопротезирование тазобедренного сустава – что нас ждет? / И.И. Шубняков, А.А. Корыткин, А.О. Денисов [и др.] // Травматология и ортопедия России. – 2025. – Т. 31, № 2. – С. 132-152. – DOI 10.17816/2311-2905-17697.
- Дефекты вертлужной области типа 3B по Paprosky: типичная картина или разнообразие вариантов? / А.Н. Коваленко, Р.М. Тихилов, А.А. Джавадов [и др.] // Травматология и ортопедия России. – 2025. – Т. 31, № 4. – С. 5–14. – DOI 10.17816/2311-2905-17736.
- Early outcomes of using custom-made augments in revision total hip arthroplasty / R.M. Tikhilov, A.A. Dzhavadov, A.S. Demin [et al.] // International Orthopaedics. – 2022. – DOI 10.1007/s00264-022-05489-9.
- Волокитина Е.А., Хабиб М.С. Эндопротезирование тазобедренного сустава при деформациях и дефектах вертлужной впадины (обзор литературы). Уральский медицинский журнал. 2018; (1): 56–63.
- Paprosky W., Sporer S., O’Rourke M.R. The treatment of pelvic discontinuity with acetabular cages. Clin Orthop Relat Res. 2006; (453): 183–187. DOI: 10.1097/01.blo.0000246530.52253.7b.
- Paprosky W.G., Perona P.G., Lawrence J.M. Acetabular defect classification and surgical reconstruction in revision arthroplasty. A 6-year follow-up evaluation. J. Arthroplasty. 1994, 9, (1): 33–44.
- Каминский А.В., Марченкова Л.О., Поздняков А.В. Ревизионное эндопротезирование тазобедренного сустава: эпидемиология, причины, факторы риска (обзор зарубежной литературы) // Вестник травматологии и ортопедии им. Н.Н. Приорова. 2015. Т. 22, № 2. С. 83–89.
- Какие особенности дефекта вертлужной впадины влияют на выбор ацетабулярного компонента при ревизионном эндопротезировании тазобедренного сустава? / Р.М. Тихилов, А.А. Джавадов, А.Н. Коваленко [и др.] // Травмато логия и ортопедия России. – 2020. – Т. 26, № 2. – С. 31–49. – DOI 10.21823/2311-2905-2020-26-2-31-49.
- Тихилов Р.М., Шубняков И.И., Денисов А.О. Классификации дефектов вертлужной впадины: дают ли они объективную картину сложности ревизионного эндопротезирования тазобедренного сустава? (критический обзор литературы и собственных наблюдений). Травматология и ортопедия России. 2019. Т. 25. (1): 122–141. DOI: 10.21823/23112905-2019-25-1-122-141
- Соглашение о единых принципах и правилах обращения медицинских изделий (изделий медицинского назначения и медицинской техники) в рамках Евразийского экономического союза» (Заключено в г. Москве 23.12.2014) (ред. от 13.02.2023).
- Анализ экономической эффективности использования индивидуальных и серийных вертлужных конструкций при ревизионном эндопротезировании тазобедренного сустава / Р.М. Тихилов, А.А. Джавадов, А.О. Денисов [и др.] // Гений ортопедии. – 2022. – Т. 28, № 2. – С. 234–240. – DOI 10.18019/1028-4427-2022-28-2-234-240.